
26. Vascular liver disease
Matthias J. Bahr
“It is impossible to explain or to understand the morbid appearances of the liver, without referring to its intimate structure, and as some points relating to this have been only lately made out, I shall commence with a short account of it.”
Georg Budd, Diseases of the Liver, 1853
Vascular liver diseases comprise a heterogeneous group of mostly rare hepatic disorders – some of them exceedingly rare.
Every single part of the hepatic vasculature may be affected, i.e., hepatic sinusoids, portal vein, hepatic artery and liver veins. The clinical presentation varies widely depending on the type of disease but also within the individual disease entities. Vascular liver diseases may present as acute disorders or chronic liver disease, as hepatocellular necrosis or cholestasis, as tumour-like lesions or portal hypertension.
The spectrum of underlying causes is wide, and in many cases multiple risk factors will result in the development of clinically significant disease (Table 1).
Table 1. Classification of predisposing factors for vascular liver disease| Hereditary disorders |
|
| Congenital or acquired malformations |
|
| Acquired cellular defects |
|
| Inflammatory disease, immune-mediated disorders |
|
| Toxicity, radiation, trauma |
Disorders of the hepatic sinusoid
Hepatic sinusoidal disease may present as luminal obstruction (i.e., sinusoidal obstruction syndrome), as luminal enlargement (i.e., peliosis hepatis) or as perisinusoidal fibrosis. Whether the latter represents a separate disease entity is debatable, as perisinusoidal fibrosis is also observed in common diseases such as steatohepatitis. Both sinusoidal obstruction syndrome as well as peliosis hepatis are not strictly confined to the hepatic sinusoids but may extend to the hepatic venous system.
Sinusoidal obstruction syndrome (Hepatic veno-occlusive disease)
Sinusoidal obstruction syndrome (SOS), also referred to as hepatic veno-occlusive disease (VOD), is a circulatory disorder primarily affecting the hepatic sinusoids. Involvement of the hepatic central veins may occur, but studies after conditioning for hematopoietic cell transplantation have demonstrated that in more than 40% of patients with SOS the hepatic venous system is not involved. The proportion of sole sinusoidal affection falls to 25% in patients with progression to severe SOS (DeLeve 2009).
Pathophysiology
Sinusoidal obstruction syndrome may be triggered by a variety of factors (Valla 2016). By far the most common cause in the Western world are myeloablative regimens in preparation for hematopoietic stem cell transplantation (HSCTx), particularly when the transplant is for a malignancy. Historically, the proportion of patients with SOS after HSCTx varied from the single-digit percentage range up to 50% if highly toxic regimens were chosen. Currently, rates between 8% and 14% are reported (Mohty 2015, Richardson 2013). Apart from conditioning regimens for HSCTx (high-dose chemotherapy plus total body irradiation), other drugs have been implicated in the development of SOS (Table 2). Among others and in addition to the intensity of the chemotherapy applied, additional risk factors appear to increase the risk for SOS: genetics, Karnofsky score, exposure to estroprogestatives in women, autologous or allogeneic type of HSCTx, prior myeloablative transplantation or preexistent liver disease (DeLeve 2009, Mohty 2016).
Originally, the syndrome was described in conjunction with the ingestion of herbal teas or foods containing pyrrolizidine alkaloids. Rarely, SOS is caused by hereditary SP110 defects also leading to immunodeficiency syndrome, VODI (Cliffe 2012). Whether immunodeficiency may give rise to infections causing secondary SOS is under dedate. In addition, MTHFR mutations are suggested as a risk factor for SOS (Efrati 2014).
Both the histopathological changes and the clinical picture of SOS were experimentally studied in a rat model using monocrotaline, a pyrrolizidine alkaloid that is directly toxic to sinusoidal endothelial cells. These experiments have confirmed the primary sinusoidal damage infrequently followed by central venous involvement (DeLeeve 1996, Mohty 2015). In addition, chemotherapy might disturb sinusoidal repair by inhibiting mobilisation of bone marrow progenitors of endothelial cells (Vion 2015).
Table 2. Conditions associated with sinusoidal obstruction syndrome
|
|
| DRUGS | |
|
|
**Reports for azathioprine-associated SOS included concurrent potential causes of SOS (modified according to DeLeve 2009, Thatishetty 2013, Tewari 2017)
Clinical presentation and diagnosis
SOS characteristically presents with weight gain (associated or not with ascites), hepatomegaly with right upper quadrant pain, and jaundice. The onset of symptoms usually occurs between day 10 and day 20 after cyclophosphamide-containing regimens but can be delayed up to 1 month after conditioning therapy if other therapies are used.
Primarily, SOS is a clinical diagnosis with the following characteristics: (1) hepatotoxic conditioning regimen for HSCTx with an appropriate temporal relation to the development of clinical signs and symptoms, (2) weight gain & hepatic pain & jaundice and, (3) negative work-up for other causes (Dignan 2013, Bajwa 2017). In patients meeting these criteria, diagnosis can be made with reasonable certainty and solely based on clinical judgement. Differential diagnoses comprise cholestatic jaundice due to sepsis, drug-induced cholestasis, fluid overload due to renal failure or congestive heart failure, liver involvement by viral or fungal infections, and acute graft-versus-host disease.
However, in up to 20% of patients the diagnosis of SOS cannot reliably be made on clinical grounds (McDonald 1993 & 2004). This has promoted the development of scoring systems such as the Seattle or the Baltimore Criteria (Jones 1987; McDonald 1993) (Table 3). However, up to 50% of patients not meeting the Baltimore criteria may exhibit histological features of SOS (Shulman 1994). Measurement of various biomarkers was suggested as indicator and follow-up marker of SOS (e.g. von Willebrand factor, thrombomodulin, E-selectin, sICAM1, PAI-1). Their use, however, is still regarded as experimental (Dignan 2013, Bajwa 2017). In 2016 the European Society for Blood and Marrow Transplantation revised the criteria for diagnosis and severity (Table 4). Taking into account that the paediatric population significantly differs from adults, separate criteria were recently established for children (Corbacioglu 2018).
Table 3. Diagnosis of sinusoidal obstruction syndrome after HSCTx| Seattle criteria (McDonald 1993) | Baltimore criteria (Jones 1987) |
At least two of the following findings within 20 days of transplantation:*
|
Hyperbilirubinaemia >34.2 µmol/L (2 mg/dL) plus ≥2 additional criteria
|
| Classical SOS In the first 21 days after HSCT | Late onset SOS >21 Days after HSCT |
Bilirubin >34 μmol/L (2 mg/dL) and two of the following criteria must be present:
|
|
The gold standard to confirm SOS is based on the combination of hepatic histology plus measurement of the wedged hepatic venous pressure gradient (HVPG >10 mmHg, specificity >90%, PPV >85%). Both can be achieved during a single procedure via the transvenous route, especially as increased bleeding risk often precludes percutaneous liver biopsy. However, histology may be negative due to the sometimes patchy character of the disease.
Imaging techniques are used to confirm hepatomegaly or ascites and will help to rule out differential diagnoses such as biliary obstruction. A more specific sign is the finding of hepatic inflow blockage with reduced or reversed portal flow in colour Doppler ultrasound (Figure 1). In addition, attenuation of hepatic venous flow or gallbladder wall edema may be detected. Some authors suggest the use of composite ultrasound imaging scores (Lassau 2002). Though less specific, CT imaging (i.e. heterogeneous hypoattenuation and patchy enhancement in the portal venous or equilibrium phase) may be suggestive for SOS (Yang 2018).
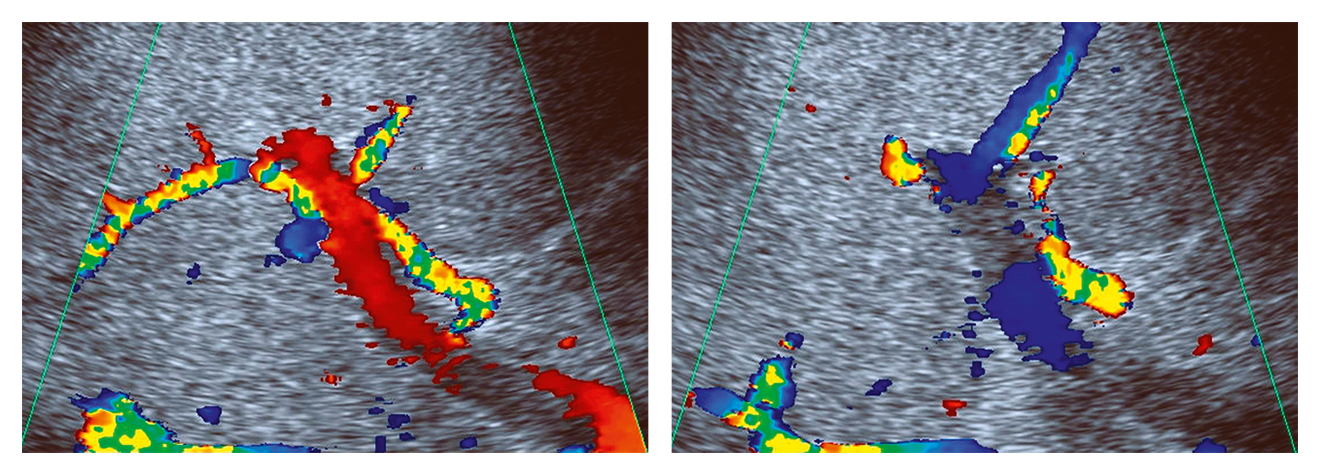 Figure 1. Doppler ultrasound in sinusoidal obstruction syndrome. Exemplary case showing undulating portal venous flow in a jaundiced patient after HSCTx
Figure 1. Doppler ultrasound in sinusoidal obstruction syndrome. Exemplary case showing undulating portal venous flow in a jaundiced patient after HSCTx
Severity of SOS varies from mild forms to rapidly progressing and eventually life-threatening disease (McDonald 1993). In patients without need for treatment of fluid excess or hepatic pain, SOS is considered mild and is associated with a self-limited course. Treatment associated with a complete remission within 100 days is considered moderate disease. If SOS does not resolve by day 100, it is categorised as severe. This classification, however, is retrospective and does not support clinical decision-making. The EBMT has proposed a modified classification system (Mohty 2016) (Table 5).
Table 5. EBMT criteria for severity of sinusoidal obstruction syndrome in adults (Mohty 2016)| Milda | Moderatea | Severe | Very severe | |
| Time since first clinical symptoms of SOS c | >7 Days | 5–7 Days | ≤4 Days | Any time |
| Bilirubin (μmol/L) | ≥34 and <51 | ≥51 and <85 | ≥85 and <136 | ≥136 |
| Bilirubin kinetics | Doubling within 48 h | |||
| Transaminases | ≤2 x normal | >2 and ≤5 x normal | >5 and ≤8 x normal | >8 x normal |
| Weight increase | <5% | ≥5% and <10% | ≥5% and <10% | ≥10% |
| Renal function | <1.2 x baseline at transplant | ≥1.2 and <1.5 x baseline at transplant | ≥1.5 and <2 x baseline at transplant | ≥2 x baseline at transplant or signs of MOD/MOFb |
b Multi-organ dysfunction must be classified as very severe
c Time between first signs/symptoms and fulfillment of SOS diagnostic criteria
Management and prognosis
Taking into account that SOS is probably under-diagnosed by solely employing clinical criteria, case fatality rates of detected SOS vary between 15 and 20% (DeLeve 2009). Apart from deep jaundice, additional signs of liver failure such as coagulopathy or hepatic encephalopathy may be missing. In contrast, systemic complications leading to multiple organ failure (renal, pulmonary) are the main reasons for death in these patients (Mohty 2015). This underlines the necessity of a closely supervised management concept. Highly toxic conditioning regimens should possibly be avoided. Recently, SOS prophylaxis using ursodeoxycholic acid was recommended (Cheuk 2015). In high-risk patients, defibrotide may be used (Dignan 2013, Mohty 2015).
Several treatments have been suggested for established SOS, e.g., thrombolysis using tPA, defibrotide or methylprednisolone (DeLeve 2009, Dignan 2013, Richardson 2013). In addition, invasive strategies such as TIPS or liver transplantation have been evaluated. Primarily, fluid management should aim to control fluid overload (using diuretics, paracentesis, hemofiltration/hemodialysis) and adequate oxygenation should be provided (Mahadeo 2017, Ovchinsky 2018). Thrombolysis has not proved successful and was associated with severe complications. Defibrotide, a mixture of single-stranded oligodeoxyribonucleotides derived from porcine intestinal mucosa, works as an endothelial protective agent (Palomo 2016). Defibrotide was successfully tested in phase II and III trials both in paediatric and adult settings (Richardson 2010, Corbacioglu 2012, Richardson 2016). This compound can also be used in multiple organ failure without substantially increasing the bleeding risk. However, current data support defibrotide use as soon as SOS is diagnosed. Methylprednisolone may be considered as additional therapy (Dignan 2013).
Unlike Budd-Chiari syndrome, decompression of portal hypertension using TIPS does not improve SOS. For patients with favourable prognosis of the underlying hematopoietic disorder after HSCTx, liver transplant might possibly be considered.
Peliosis hepatis
Peliosis hepatis is a rare and potentially reversible disorder characterised by single or multiple blood-filled cystic cavities within the hepatic tissue. Whether it is related to nonobstructive sinusoidal dilatation is currently unclear (Marzano 2015). Prevalence of peliosis hepatis may vary between 0.03% in HIV infection, 0.2% in pulmonary tuberculosis and up to 20% after renal transplantation. There is no favoured localisation of the peliotic lesions. It may occur at all ages, including a fetal form. The size ranges from submillimetres to centimetres but rarely exceeds 3 cm. The histopathological appearance may show a missing endothelial cell lining with hepatocytes directly serving as boundary (parenchymal type). Alternatively, the endothelium may be preserved but the hepatic sinusoids appear dilated. The aneurysmal dilation may extend to the central vein (phlebectatic type) (Yanoff 1964, Tsokos 2005).
Pathophysiology
Several risk factors have been suggested as promoters of peliosis hepatis, e.g., infections, drugs or malignant disorders (Table 6). However, the exact pathogenesis of peliosis is still unclear. Histology suggests endothelial damage leading to destruction of the endothelial lining. Other hypotheses favour an increased sinusoidal pressure resulting in the widening of the sinusoidal lumen with consecutive destruction of the sinusoidal endothelium or primary hepatocellular necrosis replaced by blood-filled cystic lesions. Fibrotic changes and even liver cirrhosis as well as regenerative nodules may be found, but it is unclear whether these features are directly linked to peliosis hepatis or whether they are just coincidental.
Table 6. Risk factors reported with peliosis hepatis| Infections |
|
| Drugs, toxins |
|
| Malignant and benign tumours |
|
| Inflammatory disease |
|
| Miscellaneous |
|
Clinical presentation and diagnosis
Peliosis hepatis is mostly asymptomatic and incidentally detected by hepatic imaging. Rarely, the peliotic cysts may rupture leading to intrahepatic or intraabdominal hemorrhage. Individual cases with overt liver disease have been reported, characterised by hepatomegaly, jaundice, ascites, portal hypertension and liver failure. Extrahepatic manifestations may be found in organs of the mononuclear phagocytic system (e.g., spleen, lymph nodes, bone marrow) but also in the lungs, kidneys, parathyroid or adrenal glands, or other parts of the gastrointestinal tract.
Usually, peliosis hepatis is easily detected by imaging techniques (Ronot 2016). However, discrimination between peliosis and other benign or malignant lesions may turn difficult. Peliotic lesions miss a mass effect on the adjacent hepatic vasculature. Blood flow within the lesion is slow, resulting in a hypodense appearance after contrast application in CT. However, in some patients a ring-like accumulation of contrast media may be present. Using MRI, low intensity is seen in T1-weighted images while T2-weighted images show a high signal (Iannaccone 2006). In contrast-enhanced ultrasound (CEUS) both centrifugal as well centripetal contrast filling might be detected, in some cases even tumour-like behaviour occurs (Schuldes 2011). Though imaging techniques may assist the diagnosis of peliosis hepatis, a liver biopsy is often needed for final confirmation. Wedged hepatic venography may also be diagnostic, but its use needs strong suspicion.
Management and prognosis
Typically, peliosis hepatis will not progress to symptomatic disease. In these patients management has to concentrate on the identification and, if required, treatment of the underlying disease. Causal treatment is the therapeutic mainstay mostly leading to regression of the peliotic lesions. Individual cases may require surgery if the risk of cyst rupture and consecutive bleeding is estimated to be high. If liver failure or portal hypertension dominate the clinical picture liver transplantation might be considered provided aetiology does not pose a contraindication.
Disorders of the hepatic artery
Pathologies involving the hepatic artery may lead to different clinical pictures (Table 7, Figure 2).
Occlusion of the arterial lumen results in ischaemia of the supplied tissue. Though gross hepatocellular necrosis may follow, such as in ischemic hepatitis, preserved portal venous oxygen supply often prevents the most devastating damage. In contrast to the hepatic parenchyma, the biliary system is exclusively supplied arterially and, therefore, more susceptible to ischemic damage. Clinically, this may present as an elevation of cholestasis-associated liver enzymes (i.e., gamma GT, alkaline phosphatase). In more severe cases, structural damage to bile ducts may be irreversible (i.e., ischemic cholangiopathy). Especially after orthotopic liver transplantation ischaemia type biliary lesions (ITBL) still pose a major challenge for clinical management.
Table 7. Aetiology of hepatic artery disease| Obstruction or destruction of the hepatic artery |
|
| Aneurysms |
|
| Shunts |
|
Apart from sequelae due to hepatic ischaemia, hepatic artery disease may present either as an aneurysm or as a shunt. Aneurysms of the hepatic artery are often detected incidentally by imaging. In the majority, they are asymptomatic but abdominal pain or – in rare cases – obstructive jaundice may develop. In about 20% of cases multiple aneurysms are present. Males are more often affected than women. The risk of rupture and subsequent hemorrhage is high and may reach up to 80% depending on the size of the aneurysm. Therefore, either radiological intervention or surgery needs to be evaluated (Hulsberg 2011, Christie 2011).
In contrast to aneurysms, shunts involving the hepatic artery are predominantly symptomatic. The spectrum of symptoms is wide including abdominal pain, portal hypertension or signs of high-output heart failure. The therapeutic approach has to be individualised including radiological interventions or surgical procedures.
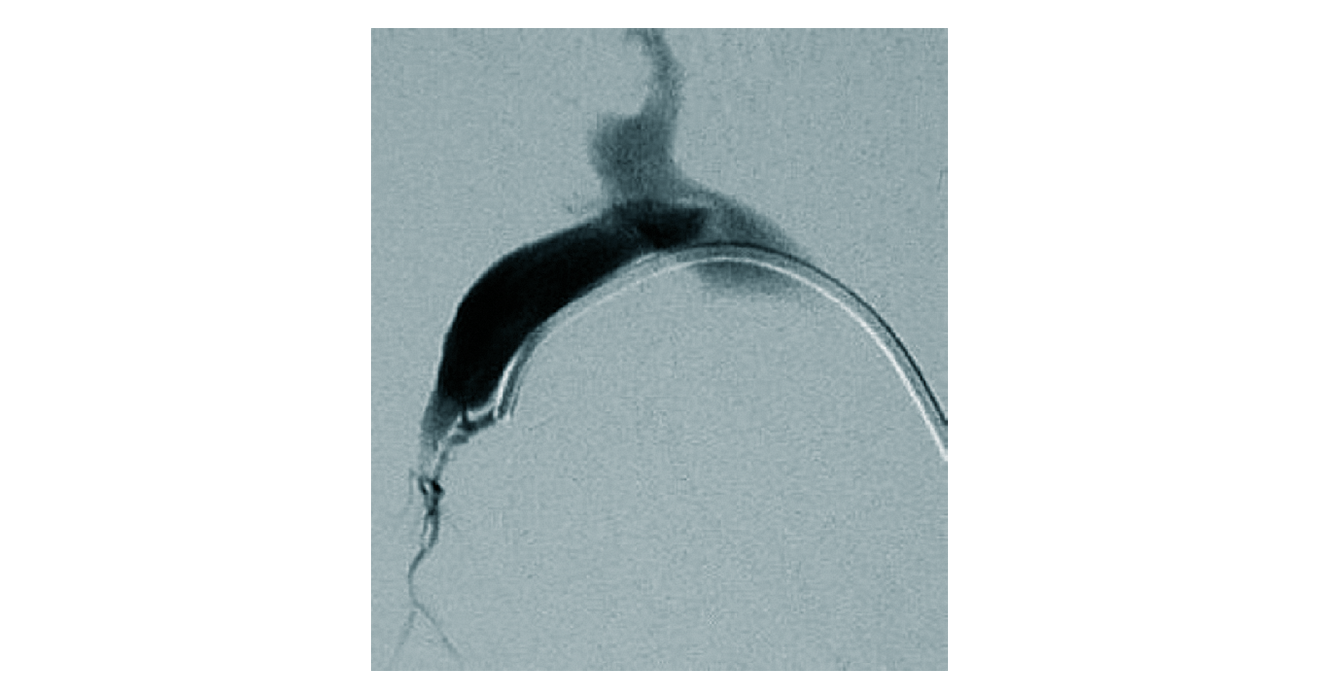 Figure 2. Spontaneous arterioportal shunt. Angiography in a patient with non-cirrhotic portal hypertension. A small arterioportal shunt is detected by superselective catheterisation
Figure 2. Spontaneous arterioportal shunt. Angiography in a patient with non-cirrhotic portal hypertension. A small arterioportal shunt is detected by superselective catheterisation
Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome)
Hereditary hemorrhagic telangiectasia (HHT) is a highly penetrant, autosomal dominant disease showing a heterozygous prevalence between 1:5,000 and 1:8,000. It is characterised by progressive and multivisceral development of arteriovenous malformations (Govani 2009, Garg 2014, Arthur 2015).
Mutations in several genes interacting with transforming growth factor (TGF)-β receptor have been identified in HHT. According to the genes involved, different subtypes can be discriminated:
- HHT 1 (ENG coding for endoglin, chromosome 9q33-q34.1),
- HHT 2 (ACVRL1 coding for activin A receptor type II-like kinase ALK-1, chromosome 12q11-q14),
- HHT 3 (gene not yet identified, chromosome 5q31.3-q32),
- HHT 4 (gene not yet identified, chromosome 7p14),
- HHT 5 (HHT5 coding for GDF-2, also known as BMP-9, chromosome 10q11.22),
- Juvenile polyposis/HHT (SMAD4, chromosome 18q21.1).
Liver involvement may be found in all subtypes but appears to be more frequent in HHT 2. Though hereditary, HHT is characterised by marked intrafamilial variation. Recently, the first case of tissue-specific mosaicism was reported (McDonald 2018).
Clinical presentation and diagnosis
HHT is a multivisceral disease. Apart from the nasopharnyx and the gastrointestinal tract, central nervous (~10%), pulmonary (~50%) and hepatic involvement occur at high frequency. Accordingly, the spectrum of clinical disease is wide, e.g., anaemia, seizures, subarachnoid hemorrhage, paraplegia, transient ischemic attacks/stroke, dyspnea, cyanosis, polycythaemia, abdominal pain and hepatic abscesses. Symptoms develop progressively throughout life. Telangiectasias appear before the age of 20 in half, before 40 in two-thirds of the patients. Thereafter it takes one or two decades for the development of significant bleeding or symptomatic visceral involvement (Plauchu 1989, Govani 2009, Arthur 2015). Overall, life expectancy of patients suffering from HHT is two decades less than in the general population (Droege 2018).
The proportion of hepatic involvement in HHT reaches up to 75%. Hepatic malformations appear more common in females. However, less than 20% of patients with hepatic involvement are symptomatic (Singh 2014). The clinical picture of liver involvement in HHT depends on the predominant type of malformation (i.e., arterioportal vs. arteriovenous shunts). Arteriovenous malformations increase cardiac output. In individual cases up to 20 L/min may be reached. These patients suffer from high output cardiac failure. In addition, symptoms of a mesenteric steal syndrome (e.g., postprandial abdominal pain) and complications of biliary ischaemia (e.g., biliary abscesses) may occur. As a consequence of ischaemia, nodular regeneration of the liver develops (HHT-associated pseudocirrhosis). Arterioportal malformations will cause portal hypertension (Buscarini 2006, Garcia-Tsao 2000).
Diagnosis of HHT is made using the Curaçao criteria, 3 of 4 of which need to be fulfilled (Shovlin 2000, Faughnan 2011):
- recurrent spontaneous epistaxis,
- telangiectasias, multiple and in typical localization,
- positive family history,
- visceral arteriovenous malformations (lung, liver, brain, spine).
| Major criteria |
|
| Minor criteria |
|
| Facultative findings |
|
Current guidelines do not endorse routine screening for hepatic vascular malformations. Recently, a diagnostic score involving age, gender, hemoglobin and alkaline phosphatase was presented to identify patients at risk for significant liver disease (Singh 2014). However, using Doppler ultrasound, screening is performed with high sensitivity and specificity (Table 8) (Caselitz 2003). If hepatic involvement is confirmed, cardiac output should be estimated (e.g., via echocardiography). Furthermore, screening at regular intervals is advised to detect complications such as development of portal hypertension or biliary lesions.
Management of hepatic involvement in HHT
Currently, no established medical therapy for HHT exists. In chronic GI bleeding the use of hormonal therapy (estrogen-progesterone preparations, danocrine), antifibrinolytics (aminocaproic acid, tranexamic acid) and other experimental drugs (tamoxifen, interferon, thalidomide, sirolimus) were suggested (Ardelean 2015, Faughnan 2011). However, no data supports the use of these drugs to treat hepatic vascular malformations. A phase 2 trial evaluated bevacizumab to treat liver involvement in HHT (Dupuis-Girod 2012). Significant improvements in cardiac output, epistaxis and SF-36 scores were achieved. However, long-term effects, dosing and necessity of maintenance therapy are still unclear (Ardelean 2015, Chavan 2017). Registry data comparing thalidomide and bevacizumab show positive effects on transfusion dependency, GI bleeding and epistaxis for both drugs while only bevacizumab was helpful in treating vascular malformations (Buscarini 2019). Single cases using kinase inhibition (i.e., sunitinib, nintedanib) were reported, but still have to be regarded experimental.
Limited data exist for the use of hepatic artery embolisation and liver transplantation (Buscarini 2006, Chavan 2013, Felli 2017). Due to the invasiveness and complication rates of these approaches only patients with moderate to severe symptoms should be regarded as candidates for interventional therapy. Hepatic artery embolisation can be used to reduce shunt flow in patients with arteriovenous hepatic shunts leading to significant reduction of cardiac output and improvement of associated symptoms. However, complications such as hepatic and biliary necrosis or acute cholecystitis have been described. Success of hepatic artery embolisation very much depends on adequate patient selection. Current guidelines do not endorse general use of embolisation outside experienced centres but do favour liver transplantation in advanced hepatic involvement of HHT.
Disorders of the portal vein
Portal vein thrombosis is a common disease located within the main portal vein and its larger branches. Additionally, rare affections of the medium-sized and preterminal portal vein branches have been identified. The nomenclature for the latter has been inconsistent (e.g., obliterative portal venopathy, hepatoportal sclerosis, idiopathic portal hypertension, nodular regenerative hyperplasia). Recently, the term idiopathic non-cirrhotic portal hypertension was established replacing and incorporating the different previously decribed subtypes (EASL 2016).
Portal vein thrombosis
Portal vein thrombosis (PVT) is the most frequent disorder affecting the hepatic vasculature. Autopsy studies report a prevalence range between 0.05% and 0.5%. In compensated cirrhosis PVT may be found in 1% of cases, while prevalence between 8% and 26% are reported for decompensated cirrhosis.
PVT is of heterogeneous aetiology. It is promoted by both local and systemic risk factors (Tables 9 & 10). In about 20 to 30% of patients a local risk factor can be identified. Systemic risk factors are found in 50-70% (DeLeve 2009, Plessier 2010). Recently, central obesity was identified as a major risk factor for idiopathic PVT (Bureau 2016).
Table 9. Local risk factors for portal vein thrombosis| Malignancy |
|
| Focal inflammation |
|
| Portal venous injury |
|
| Vascular haemodynamics |
|
Clinical presentation and diagnosis
Portal vein thrombosis may present as acute or chronic disease, representing successive stages of disease progression. As management depends on the PVT subtype, non-cirrhotic, non-malignant PVT needs to be regarded separately from (a) thrombi resulting from slowed portal venous flow in liver cirrhosis, (b) malignant thrombi resulting from tumours invading the portal venous circulation, and (c) septic thrombi also known as acute pylephlebitis (DeLeve 2009, Plessier 2010). A classification focusing on anatomico-functional aspects has found wide acceptance (Table 10) (Sarin 2016).
Table 10. Sarin classification of portal vein thrombosis (Sarin 2016)| Site of PVT | Type 1: trunk only Type 2: branch only: 2a, one branch; 2b, both branches Type 3: trunk and branches |
| Extent of PV system occlusion | S: splenic vein M: mesenteric vein SM: both |
| Degree of portal venous system occlusion | O: occlusive, no visible flow in PV lumen on imaging/Doppler study NO: non-occlusive, flow visible in PV lumen on imaging/Doppler study |
| Duration and presentation | R: recent (previously patent PV, hyperdense thrombus, absent/limited collaterals, dilated PV at the site of occlusion) Ch: chronic (previously diagnosed PVT, no hyperdense thrombus, portal cavernoma, portal hypertension) As: asymptomatic S: symptomatic |
| Type of underlying liver disease | Cirrhotic Non-cirrhotic Hepatobiliary malignancy Local malignancy Post-transplant Associated conditions |
The typical clinical presentation of acute PVT includes abdominal or lumbar pain of sudden onset or progressing over a few days. Depending on the extent of the thrombosis the pain may be severe and colicky. The diminished mesenteric outflow leads to intestinal congestion. Paralytic ileus may develop. Moderate distension of the abdomen is common. However, peritoneal signs are usually absent unless intestinal infarction develops. Fever and a marked systemic inflammatory response may develop even without systemic infection. This is accompanied by elevated laboratory markers of inflammation. In contrast, liver function – apart from intermittent elevation of aminotransferases – is usually not substantially affected by acute PVT unless significant liver damage pre-exists. Clinical features should improve within 5-7 days. Otherwise transmural intestinal ischaemia has to be suspected.
Pylephlebitis often develops secondary to a primary site of infection (e.g. pancreatitis, diverticulitis). It is characterised by high, spiking fever with chills, a painful liver, and sometimes shock. Blood cultures should be taken (often Bacteroides spp., E. coli ± other enteric species). Infected thrombi give rise to hepatic microabscesses (Kanellopoulou 2010, Choudhry 2016).
Cases without resolution of acute portal vein thrombosis progress to the chronic stage. The obstructed portal vein is replaced by collateral veins bridging the thrombotic part, known as portal cavernoma (also addressed as Extra Hepatic Portal Venous Obstruction, EHPVO). There is wide variation in the clinical picture of portal cavernoma. It may rarely lead to obstruction of the extrahepatic bile ducts (i.e., portal cholangiopathy/biliopathy, portal cavernoma cholangiopathy), which may be associated with marked jaundice (Dhiman 2014, Khuroo 2016). However, the leading symptom of chronic PVT are the facets of portal hypertension (e.g., portosystemic collaterals such as gastric or oesophageal varices). As liver function is usually not impaired, complications such as hepatic encephalopathy or ascites are substantially less frequent than in liver cirrhosis. Hepatopulmonary syndrome may be found in up to 10% of patients.
PVT is a common complication of liver cirrhosis with an increasing prevalence in more severe disease stages. It needs to be discriminated from portal venous obstruction caused by hepatocellular carcinoma. Pathophysiologically, PVT in cirrhosis arises as a consequence of reduced hepatic inflow leading to diminished flow velocity and eventually stasis within the portal vein. Therefore, thrombi are often partial and development of portal cavernoma is rather unusual. The use of non-selective beta-blockers (NSBB) in cirrhosis may increase the risk of PVT development by more than 4-fold (Xu 2019). In patients with cirrhosis, a newly developed ascites or significant worsening of existing ascites should trigger the search for PVT.
Both acute PVT and portal cavernoma are easily detected using sonography, CT or MR imaging. Acute PVT presents as intraluminal hyperechoic material in ultrasound, while Doppler imaging demonstrates a lack of blood flow (Figure 3). Using contrast-enhanced ultrasound (CEUS), vascularisation of the thrombus may be used to identify malignant thrombi. As PVT may extend to the mesenteric or splenic veins, thorough assessment of the splanchnic tributaries is mandatory. For detailed assessment of thrombus extension, CT or MR angiography are more sensitive than Doppler sonography. Portal cavernoma presents as serpiginous vessel structures, while the main portal vein or its branches are not visible. As a compensatory mechanism hepatic arteries are usually enlarged. Depending on the individual location and appearance of portal cavernoma it may be mistaken as part of the surrounding organs or as tumour.
Management and prognosis
In acute PVT, recanalisation of the obstructed veins should be aspired. Causal factors require correction where possible. Spontaneous recanalisation without anticoagulation occurs infrequently (<10%). Therefore, anticoagulation is the most commonly used therapeutic strategy to reopen the obstructed portal vein. Though no controlled studies exist, prospective data suggest success rates between 25% and 80%. Response increases if neither the splenic vein is involved nor ascites is detectable. Anticoagulation should be initiated as early as possible – delay might be associated with treatment failure. Major complications are reported in less than 5% of treated patients (DeLeve 2009, Plessier 2010, Hall 2011). Direct oral anticoagulants are not used as routine anticoagulation yet, but may represent a more convenient anticoagulation alternative for PVT management in future practice (Wu 2019). Depending on whether a transient or a persistent risk factor has facilitated PVT development, anticoagulation should be maintained for 6 months or long-term, respectively (EASL 2016).
Experience with other treatment modalities is limited (e.g., systemic/local thrombolysis, surgical thrombectomy, transjugular intrahepatic portosystemic stent [TIPS]). Systemic thrombolysis appears largely ineffective. Although performed successfully in some centres, major procedure-related complications and even death have been reported for local thrombolysis. A recent meta-analysis attested that TIPS placement is technically highly feasible, effective and safe (Rodrigues 2018). Emergency surgical intervention is indicated in suspected intestinal infarction. In these cases, surgical thrombectomy can be performed.
If treatment is initiated early in acute PVT the outcome is favourable. Symptoms may sometimes disappear within hours after start of therapy and portal hypertension rarely develops. Overall mortality is well below 10% (DeLeve 2009, Plessier 2010).
In patients with portal cavernoma, prevention of gastrointestinal bleeding due to portal hypertension is a main focus of therapy (Chaudhary 2013). The use of non-selective beta-blockers is incompletely evaluated in portal cavernoma. However, an approach similar to portal hypertension in liver cirrhosis is supported by current guidelines and appears to improve prognosis (DeLeve 2009). Due to the variable genesis of PVT, individual assessment for risk of recurrence of thrombosis and risk of bleeding should be performed. Although data is scarce, anticoagulation seems to be favourable for most patients.
The therapeutic approach in patients with PVT associated with liver cirrhosis has to be regarded separately. Whether PVT increases mortality in patients with cirrhosis remains unclear (Berry 2015, Cool 2019). On the other hand, anticoagulation is safe both in the prophylactic as well as in the therapeutic setting (Villa 2012, Delgado 2012). Use of enoxaparin as primary prophylaxis completely prevented the development of PVT. In subacute PVT, anticoagulation (using either vitamin K antagonists or LMWH) achieved complete recanalisation in nearly half of the patients, while at least partial response was seen in 2/3 of cases. Similar results are achieved using DOAC (De Gottardi 2017). Interventional therapy using TIPS appears even more effective showing complete response in 57% and at least partial response in all patients (Luca 2011, Rössle 2014). Preliminary data suggest that systemic thrombolysis is feasible (De Santis 2010). As PVT does not change the clinical course of liver cirrhosis, primary treatment candidates are patients on the waiting list for liver transplantation.
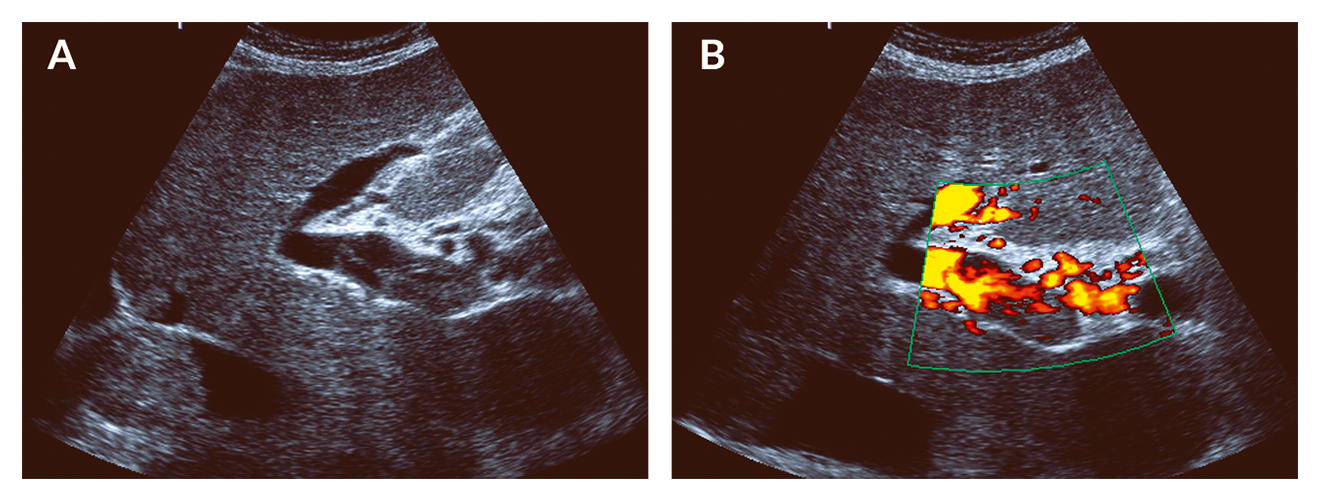 Figure 3. Acute portal vein thrombosis. Ultrasound of patient with acute PVT. (A) Hyperechoic material is located within the main portal vein. (B) Using the power mode for flow detection, blood flow is limited to those parts of the portal vein without hyperechoic material
Figure 3. Acute portal vein thrombosis. Ultrasound of patient with acute PVT. (A) Hyperechoic material is located within the main portal vein. (B) Using the power mode for flow detection, blood flow is limited to those parts of the portal vein without hyperechoic material
Malignant PVT resulting from hepatocellular carcinoma should not lead to therapeutic nihilism. While systemic therapy (e.g. sorafenib) is the recommended strategy in Western countries, experience from Asia favours resection to TACE or conservative treatment (Lu 2019, Zhang 2019). Portal vein stenting has been reported for malignant PVT, however, the effect on relevant end-points is unclear.
If pylephlebitis is suspected antibiotic therapy must be commenced immediately. Additional anticoagulation appears to improve outcomes in this setting (Naymagon 2020).
Idiopathic non-cirrhotic portal hypertension (INCPH)
The term INCPH was introduced to replace an ambiguous nomenclature including hepatoportal sclerosis, non-cirrhotic portal fibrosis, idiopathic portal hypertension, incomplete septal cirrhosis, nodular regenerative hyperplasia and obliterative portal venopathy (EASL 2016). Most recently, an even more comprehensive nomenclature was proposed, i.e. porto-sinusoidal vascular disease (De Gottardi 2019).
The histopathological correlate is an affection of the medium-sized and preterminal portal venous branches generating different morphological features (Guido 2019):
(a) Occlusion of the portal venous branches induces hypotrophy of the supplied tissue. As a compensatory reaction, growth of appropriately perfused liver tissue gives rise to the development of regenerative nodules. This combination of hypotrophic and hypertrophic liver tissue without signs of fibrosis is the equivalent of nodular regenerative hyperplasia (Wanless 1990).
(b) As a second type of reaction, portal veins are not just destroyed but replaced by filiform fibrotic strands penetrating the hepatic tissue. These fibrotic strands are strictly confined to the portal tracts and do not form fibrotic septae (Aggarwal 2013, Nakanuma 2001). This feature is equivalent to hepatoportal sclerosis.
Both histological features may exist side by side.
Nodular regenerative hyperplasia is found in 14-27% of cases with non-cirrhotic portal hypertension (Naber 1991, Nakanuma 1996). In autopsy studies the prevalence is 3.1/100,000, one third of which are associated with portal hypertension (Colina 1989). Hepatoportal sclerosis less frequently described in the Western world but is more common in Asia (e.g., India, Japan).
A number of associated pathologies have been suggested to promote INCPH: Immune and hematologic disorders, e.g., rheumatoid arthritis, Felty’s syndrome, other connective tissue disorders, CVID, HIV infection, myeloproliferative and lymphoproliferative disease. INCPH has been described in infective endocarditis, inflammatory bowel disease and after kidney transplantation. Furthermore, it may occur in conjunction with chemotherapy, HAART, other drugs and after toxin exposure (e.g., arsenic, vinyl chloride). Also, a hereditary component is discussed (Albuquerque 2013, Ghabril 2014, Hartleb 2011, Matsumoto 2000, Sarin 2007, Schouten 2011, Schouten 2015, Vilarinho 2016).
Clinically, INCPH presents with complications of portal hypertension. Liver function is usually not significantly impaired, although individual cases with liver failure and liver transplantation have been described. The prognosis depends on the underlying disorder and on the control of portal hypertension (Ataide 2013, Blendis 1978, Dumortier 2001, Naber 1990, Sarin 2007, Schouten 2015, Siramolpiwat 2014). TIPS has proven an effective measure in INCPH (Bissonnette 2016).
EASL guidelines have suggested all of the following 5 criteria to be fulfilled for the diagnosis of INCPH: a) Clinical signs of portal hypertension, b) exclusion of cirrhosis on liver biopsy, c) exclusion of chronic liver disease causing cirrhosis or noncirrhotic portal hypertension (chronic viral hepatitis B/C, NASH/ASH, autoimmune hepatitis, hereditary haemochromatosis, Wilson‘s disease, primary biliary cholangitis), 4) exclusion of conditions causing non-cirrhotic portal hypertension (congenital liver fibrosis, sarcoidosis, schistosomiasis), 5) patent portal and hepatic veins.
The above criteria point to the importance of liver biopsy for the diagnosis of INCPH. However, interobserver agreement in histology evaluation is variable (Jharab 2015). Even more, histological features of INCPH may be found in up to 10% of the general population (Zuo 2017). In imaging studies, differentiation between nodular regenerative hyperplasia and cirrhosis may be impossible. In ultrasound, “atoll-like lesions” have been described as a characteristic imaging feature (Caturelli 2011). A recent paper pointed to value of non-cirrhotic transient elastography results for the diagnosis of INCPH (Seijo 2012).
Disorders of the hepatic veins
Budd-Chiari syndrome is the only defined entity of hepatic venous disease. However, other disorders such as the sinusoidal obstruction syndrome or peliosis hepatis may also affect the hepatic venous system. Furthermore, hepatic congestion due to cardiac or pericardial disease shares clinical similarities with Budd-Chiari syndrome.
Budd-Chiari syndrome
Budd-Chiari syndrome (BCS) is defined as hepatic venous outflow obstruction at any level from the small hepatic veins to the junction of the inferior vena cava (IVC) and the right atrium, regardless of the cause of obstruction (Janssen 2003). Excluded by definition are obstructions caused by sinusoidal obstruction syndrome and cardiac or pericardial disorders. BCS is a rare disorder with an estimated incidence of 1 per million and a prevalence of 11 per million (Li 2019).
Pathophysiology
Obstruction of the hepatic outflow may arise from endoluminal lesions, e.g., thrombosis, webs, endophlebitis (primary BCS) or from outside the venous system by luminal invasion or by extrinsic compression, e.g., tumour, abscess, cysts (secondary BCS) (Janssen 2003).
On rare occasions, BCS originates from congenital malformations, e.g., webs or stenotic vessels (Ciesek 2010, Darwish Murad 2009). However, outflow obstruction is usually caused by thrombosis. Prevalence of thrombophilic risk factors is shown in Table 11. However, the underlying etiologies may vary in different parts of the world (Qi 2016). Thrombi are exclusively located within the hepatic veins in 49% of patients, exclusively within IVC in 2%, and as combined thrombosis of hepatic veins and IVC in 49%. In about 18% a concomitant portal vein thrombosis is identified (Darwish Murad 2009).
Obstruction of hepatic outflow leads to congestion of the drained tissue. Over time this will induce hypotrophy of affected and consecutive regenerative growth of non-affected parts of the liver. A typical area of hypertrophy is liver segment 1 (caudate lobe), favoured by its separate venous drainage into the IVC. Regenerative nodules may occasionally progress to hepatocellular carcinoma. In addition, intrahepatic collaterals may develop.
Table 11. Prevalence of thrombophilic risk factors in acute and chronic portal vein thrombosis and in primary Budd-Chiari syndrome*| Risk factor | Portal vein thrombosis | Budd-Chiari syndrome |
| Myeloproliferative neoplasms Atypical Classical | 21% – 40% 14% 17% | 40% – 50% 25% – 35% 10% – 25% |
| Paroxysmal nocturnal hemoglobinuria | 0% – 2% | 0% – 19% |
| Antiphospholipid syndrome | 6% – 19% | 4% – 25% |
| Factor V Leiden mutation | 3% – 32% | 6% – 32% |
| Factor II (prothrombin) mutation | 14% – 40% | 3% – 7% |
| Protein C deficiency | 0% – 26% | 4% – 30% |
| Protein S deficiency | 2% – 30% | 3% – 20% |
| Antithrombin deficiency | 0% – 26% | 0% – 23% |
| Plasminogen deficiency | 0% – 6% | 0% – 4% |
| Hyperhomocysteinaemia TT677 MTHFR genotype | 11% – 22% 11% – 50% | 22% – 37% 12% – 22% |
| Recent pregnancy | 6% – 40% | 6% – 12% |
| Recent oral contraceptive use | 12% – 44% | 6% – 60% |
| Behçet’s disease | 0% – 31% | 0% – 33% |
| Connective tissue disease | 4% | 10% |
Clinical presentation and diagnosis
Depending on the location of outflow obstruction, the number of vessels involved and the temporal dynamics of BCS, the clinical presentation varies between subclinical disease to light symptoms, and dramatic acute complaints which may progress to acute liver failure. The disease might present with a progressively relapsing course successively involving different hepatic veins.
Symptoms of hepatic congestion are ascites (>80% of patients), abdominal pain (>60%) and oesophageal varices (>50%). Significant disturbance of liver function is rather rare, e.g., hepatic encephalopathy (<10%), as is involvement of extrahepatic organs, e.g., hepatorenal syndrome (<10%) (Darwish Murad 2009).
In the majority of cases, diagnosis of BCS can be obtained using Doppler ultrasound. If technical difficulties obviate sonographic diagnosis, MRI is the imaging method of choice. Only in rare cases is liver biopsy or hepatic venography required to confirm the diagnosis (Janssen 2003). Ultrasound characteristics of BCS are clearly defined (Boozari 2008). They comprise specific signs such as direct visualisation of thrombi, stenosis, webs, replacement of hepatic veins by fibrotic strands or reversed flow in hepatic veins or IVC. Suggestive signs are hepatic collaterals that may be interposed between hepatic veins or may be located on the hepatic capsule. Widening of the caudate vein (>3 mm) is also regarded as suggestive for BCS. These signs serve in the diagnosis of BCS and may be accompanied by a myriad of non-specific changes (e.g., ascites, regenerative nodules, splenomegaly).
As regeneration nodules in BCS may progress to hepatocellular carcinoma, thorough imaging is mandatory. However, identification of malignant transformation may be difficult (Van Wettere 2019).
Management and prognosis
Treatment of BCS has to be adjusted to the aetiology and the severity of the clinical picture. If BCS is caused by congenital malformations such as webs, radiological interventions using balloon catheter-assisted dilation may succeed.
In case of a primary thrombotic event, anticoagulation is the mainstay of therapy (Janssen 2003, DeLeve 2009, Darwish Murad 2009, Seijo 2013, EASL 2015). However, in long-term follow-up less than half of patients will be solely treated with anticoagulation and remain free of further interventions (Seijo 2013). Therefore, interventional techniques (e.g., TIPS, recanalisation) should be evaluated early, especially in patients with moderate to severe symptoms. With the advent of TIPS, the necessity for liver transplantation in BCS has declined sharply. Success rates of TIPS – both in the short-term and in the long-term – are high (Seijo 2013, Zhang 2015). Thus, surgical procedures (e.g., surgical shunt, liver transplantation) are only rarely performed. With this approach, current data show that survival in BCS is above 70% after 5 years (Seijo 2013).
References
Aggarwal S, Fiel MI, Schiano TD. Obliterative portal venopathy: a clinical and histopathological review. Dig Dis Sci 2013;58:2767-76.
Albuquerque A, Cardoso H, Lopes J, Cipriano A, Carneiro F, Macedo G. Familial occurrence of nodular regenerative hyperplasia of the liver. Am J Gastroenterol 2013;108:150-1.
Ardelean DS, Letarte M. Anti-angiogenic therapeutic strategies in hereditary hemorrhagic telangiectasia. Front Genet 2015;6 (35):1-7.
Arthur H, Geisthoff U, Gossage JR, et al. Executive summary of the 11th HHT international scientific conference. Angiogenesis 2015;18:511-24.
Ataide EC, dos Santos IN, Martins DL, et al. Liver failure and the need for transplantation in 6 patients with hepatoportal sclerosis. Transpl Proc 2013;45:1907-9.
Bajwa RPS, Mahadeo KM, Taragin BH, et al. Consensus report by paediatric acute lung injury and sepsis investigators and paediatric blood and marrow transplantation consortium joint working committees: Supportive care guidelines for management of veno-occlusive disease in children and adolescents, part 1: Focus on investigations, prophylaxis, and specific treatment. Biol Blood Marrow Transplant 2017;23:1817-1825.
Berry K, Taylor J, Liou IW, Ioannou GN. Portal vein thrombosis is not associated with increased mortality among patients with cirrhosis. Clin Gastroenterol Hepatol 2015;13:585-93.
Bissonnette J, Garcia-Pagán JC, Albillos A, et al. Role of the transjugular intrahepatic portosystemic shunt in the management of severe complications of portal hypertension in idiopathic noncirrhotic portal hypertension. Hepatology 2016;64:224-31.
Blendis LM, Lovell D, Barnes CG, Ritland S, Cattan D, Vesin P. Oesophageal variceal bleeding in Felty’s syndrome associated with nodular regenerative hyperplasia. Ann Rheum Dis 1978;37:183-6.
Budd G. On diseases of the liver. Blanchard and Lea. Philadelphia 1853.
Buscarini E, Butella LM, Geisthoff U, et al. Safety of thalidomide and bevacizumab in patients with hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 2019;14:28.
Buscarini E, Plauchu H, Garcia Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int 2006;26:1040-6.
Bureau C, Laurent J, Robic MA, et al. Central obesity is associated with non-cirrhotic portal vein thrombosis. J Hepatol 2016;64:427-32.
Boozari B, Bahr MJ, Kubicka S, Klempnauer J, Manns MP, Gebel M. Ultrasonography in patients with Budd-Chiari syndrome – diagnostic signs and prognostic implications. J Hepatol 2008;49:572-80.
Caselitz M, Bahr MJ, Bleck JS, et al. Sonographic criteria for the diagnosis of hepatic involvement in hereditary hemorrhagic telangiectasia (HHT). Hepatology 2003;37:1139-46.
Caturelli E, Ghittoni G, Ranalli TV, Gomes VV. Nodular regenerative hyperplasia of the liver: coral atoll-like lesions on ultrasound are characteristic in predisposed patients. Br J Radiol 2011; 84:e129–e34.
Chavan A, Schumann-Binarsch S, Luthe L, et al. Systemic therapy with bevacizumab in patients with hereditary hemorrhagic telangiectasia (HHT). Vasa 2013;42:106-10.
Chavan A, Schumann-Binarsch S, Schmuck B, et al. Emerging role of bevacizumab in management of patients with symptomatic hepatic involvement in hereditary hemorrhagic telangiectasia. Am J Hematol 2017;92:E641-E647.
Chavan A, Luthe L, Gebel M, et al. Complications and clinical outcome of hepatic artery embolisation in patients with hereditary haemorrhagic telangiectasia. Eur Radiol 2013;23:951-7.
Chaudhary N, Mehrotra S, Srivastava M, Nundy S. Management of bleeding in extrahepatic portal venous obstruction. Int J Hepatol 2013;2013:784842.
Cheuk DKL, Chiang AKS, Ha SY, Chan GCF. Interventions for prophylaxis of hepatic veno-occlusive disease in people undergoing haematopoietic stem cell transplantation. Cochrane Database Syst Rev 2015;5, CD009311:1-109.
Choudhry AJ, Baghdadi YMK,Amr MA, et al. Pylephlebitis: a review of 95 cases. J Gastrointest Surg 2016;20:656-61.
Christie AB, Christie DB, Nakayama DK, Solis MM. Hepatic artery aneurysms: evolution from open to endovascular repair techniques. Am Surgeon 2011;77:608-11.
Ciesek S, Rifai K, Bahr MJ, et al. Membranous Budd-Chiari syndrome in caucasians. Scand J Gastroenterol 2010;45:226-34.
Cliffe ST, Bloch DB, Suryani S, et al. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol 2012;130:735-42.
Colina F, Alberti N, Solis JA, Martinez-Tello FJ. Diffuse nodular regenerative hyperplasia of the liver (DNRH). A clinicopathologic study of 24 cases. Liver 1989; 9:253-65.
Cool J, Rosenblatt R, Kumar S, et al. Portal vein thrombosis prevalence and associated mortality in cirrhosis in a nationally representative inpatient cohort. J Gastroenterol Hepatol 2019;34:1088-92.
Corbacioglu S, Carreras E, Ansari M, et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in paediatric patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant 2018;53:138-45.
Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet 2012;379:1301-9.
Darwish Murad S, Plessier A, Hernandez-Guerra M, et al. Aetiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009;151:167-75.
De Gottardi A, Rautou PE, Schouten J, et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol 2019;4:399-411.
De Gottardi A, Trebicka J, Klinger C, et al. Antithrombotic treatment with direct-acting oral anticoagulants (DOACs) in patients with splanchnic vein thrombosis and cirrhosis. Liver Int 2017;37:694-9.
DeLeve LD, Valla DC, Garcia-Tsao G. AASLD practice guidelines. Vascular disorders of the liver. Hepatology 2009;49:1729-64.
DeLeve LD, Wang X, Kuhlenkamp JF, Kaplowitz N. Toxicity of azathioprine and monocrotaline in murine sinusoidal endothelial cells and hepatocytes: the role of glutathione and relevance to hepatic venooclusive disease. Hepatology 1996;23:589-99.
Delgado MG, Seijo S, Yepes I, et al. Efficacy and safety of anticoagulation on patients with cirrhosis and portal vein thrombosis. Clin Gastroenterol Hepatol 2012;10:776-83.
De Santis A, Moscatelli R, Catalano C, et al. Systemic thrombolysis of portal vein thrombosis in cirrhotic patients: a pilot study. Digest Liver Dis 2010;42:451-5.
Dhiman RK, Saraswaty VA, Valla DC, et al. Portal cavernoma cholangiopathy: Consensus statement of a working party of the Indian National Association for Study of the Liver. J Clin Exp Hepatol 2014;4:S2-S14.
Dignan FL, Wynn RF, Hadzic N, et al. BCSH/BSBMT guideline: diagnosis and management of veno-occlusive disease (sinusoidal obstruction syndrome) following haematopoietic stem cell transplantation. Br J Haematol 2013;163:444-57.
Droege F, Thangavelu K, Stuck BA, et al. Life expectancy and comorbidities in patients with hereditary hemorrhagic telangiectasia. Vasc Med 2018; in press.
Dumortier J, Bizollon T, Scoazec JY, et al. Orthotopic liver transplantation for idiopathic portal hypertension: indications and outcome. Scand J Gastroenterol 2001;36:417-22.
Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 2012;307:948-55.
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Vascular diseases of the liver. J Hepatol 2016;64:179-202.
Efrati E, Zuckerman T, Ben-Ami E, Krivoy N. MTHFR C677T/A1298C genotype: a possible risk factor for liver sinusoidal obstruction syndrome. Bone Marrow Transpl 2014;49:726-727.
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. J Med Genet 2011;48:73-87.
Felli E, Addeo P, Faitot F, et al. Liver transplantation for hereditary hemorrhagic telangiectasia: A systematic review. HPB 2017;19:567-72.
Garg N, Khunger M, Gupta A, Kumar N. Optimal management of hereditary hemorrhagic telangiectasia. J Blood Med 2014;5:191-206.
Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000;343:931-6.
Ghabril M, Vuppalanchi R. Drug-induced nodular regenerative hyperplasia. Semin Liver Dis 2014;34:240-5.
Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet 2009;17:860-71.
Guido M, Alves VAF, Balabaud C et al. Histology of portal vascular changes associated with idiopathic non-cirrhotic portal hypertension: nomenclature and definition. Histopathology 2019;74:219-26.
Hall TC, Garcea G, Metcalfe M, Bilku D, Dennison AR. Management of acute non-cirrhotic and non-malignant portal vein thrombosis: a systematic review. World J Surg 2011;35:2510-20.
Hartleb M, Gutkowski K, Milkiewicz P. Nodular regenerative hyperplasia: Evolving concepts on underdiagnosed cause of portal hypertension. World J Gastroenterol 2011;17:1400-9.
Hulsberg P, de la Garza-Jordan J, Jordan R, Matusz P, Tubbs RS, Loukas M. Hepatic aneurysm: a review. Am Surg 2011;77:586-91.
Iannaccone R, Federle MP, Brancatelli G et al. Peliosis hepatis: Spectrum of imaging findings. AJR 2006;187:W43-W52.
Janssen HLA, Garcia-Pagan JC, Elias E, et al. Budd-Chiari syndrome: a review by an expert panel. J Hepatol 2003;38:364-71.
Jharap B, van Asseldonk DP, de Boer NKH, et al. Diagnosing nodular regenerative hyperplasia of the liver is thwarted by low interobserver agreement. PLoS ONE 2015;10:DOI:10.1371/journal.pone.0120299.
Jones RJ, Lee KS, Beschorner WE, et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation 1987;44:778-83.
Kanellopoulou T, Alexopoulou A, Theodossiades G, Koskinas J, Archimandritis AJ. Pylephlebitis: an overview of non-cirrhotic cases and factors related to outcome. Scand J Infect Dis 2010;42:804-11.
Khuroo MS, Rather AA, Khuroo NS, Khuroo MS. Portal biliopathy. World J Gastroenterol 2016; 22: 7973-82.
Lassau N, Auperin A, Leclere J, Bennaceur A, Valteau-Couanet D, Hartmann O. Prognostic value of doppler-ultrasonography in hepatic venoocclusive disease. Transplantation 2002;74:60-6.
Li Y, De Stefano V, Li H, et al. Epidemiology of Budd-Chiari syndrome: A systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 2019;43:468-74.
Lu J, Zhang XP, Zhong BY, et al. Management of patients with hepatocellular carcinoma and portal vein tumour thrombosis: comparing east and west. Lancet Gastroenterol Hepatol 2019;4:721-30.
Luca A, Miraglia R, Caruso S, et al. Short- and long-term effects of the transjugular intrahepatic portosystemic shunt on portal vein thrombosis in patients with cirrhosis. Gut 2011;60:846-52.
Mahadeo KM, McArthur J, Adams RH, et al. Consensus report by the paediatric acute lung injury and sepsis investigators and paediatric blood and marrow transplant consortium joint working committees on supportive care guidelines for management of veno-occlusive disease in children and adolescents: Part 2 - focus on ascites, fluid and electrolytes, renal, and transfusion issues. Biol Blood Marrow Transplant 2017;23:2023-33.
Marzano C, Cazals-Hatem D, Rautou PE, Valla DC. The significance of nonobstructive sinusoidal dilatation of the liver: Impaired portal perfusion or inflammatory reaction syndrome. Hepatology 2015;62:956-63.
Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver 2000;20:366-73.
McDonald GB, Hinds MS, Fisher LD, et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation – a cohort study of 355 patients. Ann Intern Med 1993;118:255-67.
McDonald GB. Liver disease of uncertain cause. Bone Marrow Transplant 2004;33:977-8.
McDonald J, Wooderchak-Donahue WL, Henderson K, et al. Tissue-specific mosaicism in hereditary hemorrhagic telangiectasia: Implications for genetic testing in families. Am J Med Genet 2018; in press.
Mohty M, Malard F, Abecassis M, et al. Sinusoidal obstruction syndrome/veno-occlusive disease: current situation and perspectives – A position statement from the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant 2015;50:781-9.
Mohty M, Malard F, Abecassis M, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 2016;51:906-12.
Naber AH, Van Haelst U, Yap SH. Nodular regenerative hyperplasia of the liver: an important cause of portal hypertension in non-cirrhotic patients. J Hepatol 1991;12:94-9.
Nakanuma Y, Hoso M, Sasaki M, et al. Histopathology of the liver in non-cirrhotic portal hypertension of unknown aetiology. Histopathology 1996; 28:195-204.
Nakanuma Y, Tsuneyama K, Ohbu M., Katayanagi K. Pathology and pathogenesis of idiopathic portal hypertension with an emphasis on the liver. Pathol Res Pract 2001;197:65-76.
Naymagon L, Tremblay D, Schiano T,·Mascarenhas J. The role of anticoagulation in pylephlebitis: a retrospective examination of characteristics and outcomes. J Thromb Thrombolysis 2020;49:325-31.
Ovchinsky N, Frazier W, Auletta JJ, et al. Consensus report by the paediatric acute lung injury and sepsis investigators and paediatric blood and marrow transplantation consortium joint working committees on supportive care guidelines for management of veno-occlusive disease in children and adolescents, part 3: Focus on cardiorespiratory dysfunction, infections, liver dysfunction, and delirium. Biol Blood Marrow Transplant 2018;24:204-18.
Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989;32:291-7.
Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al. Acute portal vein thrombosis unrelated to cirrhosis: A prospective multicenter follow-up study. Hepatology 2010;51:210-8.
Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood 2016;127:1719-27.
Richardson PG, Soiffer RJ, Antin JH, et al. Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant 2010;16:1005-17.
Richardson PG, Ho VT, Cutler C, Glotzbecker B, Antin JH, Soiffer R. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: Novel insights to pathogenesis, current status of treatment and future directions. Biol Blood Marrow Transplant 2013;19:S88-S90.
Richardson PG, Corbacioglu S, Ho VT, et al. Drug safety evaluation of defibrotide. Expert Opin Drug Saf 2013;12:123-36.
Richardson PG, Riches ML, Kernan NA, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016;127:1656-65.
Rodrigues SG, Sixt S, Abraldes JG, et al. Systematic review and meta-analysis: portal vein recanalisation and transjugular intrahepatic portosystemic shunt for portal vein thrombosis. Aliment Pharmacol Ther 2019;49:20-30.
Ronot M, Kerbaol A, Rautou PE, et al. Acute extrahepatic infectious or inflammatory diseases are a cause of transient mosaic pattern on CT and MR imaging related to sinusoidal dilatation of the liver. Eur Radiol 2016;26:3094-101.
Rössle M, Bausch B, Klinger C. Therapy algorithm for portal vein thrombosis in liver cirrhosis: The internist’s point of view. Viszeralmedizin 2014;30:401-8.
Qi X, Han G, Guo X, et al. The aetiology of primary Budd-Chiari syndrome - differences between the West and China. Aliment Pharmacol Ther 2016;44:1152-67.
Sarin SK, Kumar A, Chawla YK, et al. Noncirrhotic portal fibrosis/idiopathic portal hypertension: APASL recommendations for diagnosis and treatment. Hepatol Int 2007;1:398-413.
Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016;151:574-77.
Schouten JNL, Garcia-Pagan JC, Valla DC, Janssen HLA. Idiopathic noncirrhotic portal hypertension. Hepatology 2011;54:1071-81.
Schouten JNL, Verheij J, Seijo S. Idiopathic non-cirrhotic portal hypertension: a review. Orphanet J Rare Dis 2015;10(67):1-8
Schuldes M, Weickert U. Kontrastmittelsonographie bei Verdacht auf Lebermetastasen. Dtsch Med Wochenschr 2011;136:1255-6.
Seijo S, Reverter E, Miquel R, et al. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis 2012;44:855-60.
Seijo S, Plessier A, Hoekstra J, et al. Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology 2013;57:1962-8.
Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91:66-7.
Shulman HM, Fisher LB, Schoch HG, Henne KW, McDonald GB. Venoocclusive disease of the liver after marrow transplantation: histological correlates of clinical signs and symptoms. Hepatology 1994;19:1171-80.
Singh S, Swanson KL, Hathcock MA, et al. Identifying the presence of clinically significant hepatic involvement in hereditary haemorrhagic telangiectasia using a simple clinical scoring index. J Hepatol 2014:61;124-31.
Siramolpiwat S, Seijo S, Miquel R, et al. Idiopathic portal hypertension: natural history and long-term outcome. Hepatology 2014;59:2276-85.
Tewari P, Wallis W, Kebriaei P. Manifestations and management of veno-occlusive disease/sinusoidal obstruction syndrome in the era of contemporary therapies. Clin Adv Hematol Oncol 2017;15:130-9.
Thatishetty AV, Agresti N, O’Brien CB. Chemotherapy-induced hepatotoxicity. Clin Liver Dis 2013;17:671-86.
Tsokos M, Erbersdopler A. Pathology of peliosis. Forensic Sci Int 2005;149:25-33.
Van Wettere M, Purcell Y, Bruno O, et al. Low specificity of washout to diagnose hepatocellular carcinoma in nodules showing arterial hyperenhancement in patients with Budd-Chiari syndrome. J Hepatol 2019;70:1123-32.
Valla DC, Cazals-Hatem D. Sinusoidal obstruction syndrome. Clin Res Hepatol Gastroenterol 2016;40:378-85.
Vilarinho S, Sari S, Yilmaz G, et al. Recurrent recessive mutation in deoxyguanosine kinase causes idiopathic noncirrhotic portal hypertension. Hepatology 2016;63:1977-86.
Villa E, Cammà C, Marietta M, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012;143:1253-60.
Vion AC, Rautou PE, Durand F, Boulanger CM, Valla DC. Interplay of inflammation and endothelial dysfunction in bone marrow transplantation: Focus on hepatic veno-occlusive disease. Semin Thromb Hemost 2015;41:629-43.
Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology 1990;11:787-97.
Wu M, SchusterM, Tadros M. Update on management of portal vein thrombosis and the role of novel anticoagulants. J Clin Transl Hepatol 2019;7:154-64.
Xu X, GuoX, De Stefano V, et al. Nonselective beta‑blockers and development of portal vein thrombosis in liver cirrhosis: a systematic review and meta‑analysis. Hepatol Int 2019;13:468-81.
Yang S, Wu J, Lei S. CT features of hepatic venoocclusive disease: A meta-analysis. Acad Radiol 2018;25:328-37.
Yanoff M, Rawson AJ. Peliosis hepatis. An anatomic study with demonstration of two variities. Arch Pathol 1964;77:159-65.
Zhang F, Wang C, Li Y. The outcomes of interventional treatment for Budd-Chiari syndrome: systematic review and meta-analysis. Abdom Imaging 2015;40:601-8.
Zhang ZY, Dong KS, Zhang EL, et al. Resection might be a meaningful choice for hepatocellular carcinoma with portal vein thrombosis: A systematic review and meta-analysis. Medicine 2019;98:e18362.
Zuo C, Chumbalkar V, Ells PF, et al. Prevalence of histological features of idiopathic noncirrhotic portal hypertension in general population: A retrospective study of incidental liver biopsies. Hepatol Int 2017;11:452-60.