
25. Alcoholic hepatitis
Claus Niederau
Health and social problems due to alcohol overconsumption
Mortality due to alcohol overconsumption is high, in particular among young men (Mokdad 2000). Alcohol overconsumption not only increases the risk for liver disease but is also responsible for malignancies, accidents, violence, and social problems (Bellentani 1997, Vaillant 1995). Alcohol consumption in excess of 20–30 g for women and 40–60 g for men per day markedly increases the risk for liver disease (Becker 1996, Lucey 2008). However, liver cirrhosis is seen only in a minority of subjects with high alcohol consumption; less than 10% of subjects who drink more than 120 g of alcohol daily have cirrhosis (Bellentani 1997). In addition to the level of alcohol consumption, various other factors such as sex, other genetic characteristics and comorbidities contribute to the risk for liver disease (Nishigushi 1991, Becker 1996, Bellentani 1997, McCollough 1998, de Alwis 2007, Lucey 2009).
Alcoholic liver disease is the most prevalent cause of advanced liver disease in Europe (EASL 2012). Excessive alcohol use is also a major cause of preventable liver disease worldwide. However, despite its significant health burden, this is an area with limited research. Per capita alcohol consumption is closely associated with mortality from liver disease across countries (EASL 2012).
Excessive alcohol use and alcohol dependence may be seen as different sides of the same coin (EASL 2012). The WHO uses the terms hazardous and harmful alcohol use instead of alcohol abuse. A binge drinking pattern is becoming increasingly prevalent, mainly among young individuals, but little is known about its impact on liver disease (EASL 2012).
Quantity-frequency questionnaires and retrospective diaries can be used to calculate drinking habits. A good alternative to using quantity alcohol assessments are instruments to screen for risky drinking and alcohol dependence. Among these tools, the AUDIT (Alcohol Use Disorders Inventory Test) (Gual 2002) remains the ‘gold standard’ (EASL 2012). Brief motivational interventions should be routinely used in the medical management of alcohol use disorders (University of Sheffield 2009, Kaner 2009, EASL 2012).
Prevention of harmful alcohol use
- EASL guidelines make strong statements on preventive measures against excessive alcohol use (EASL 2012) which however still lack broader political and public support.
- Any evidence-based policy in Europe needs to implement preventive measures to reduce alcohol consumption at the population level.
- Excess alcohol consumption may need to be addressed and controlled using pricing-based policies (i.e., special taxes and tariffs, similar to cigarettes).
- Restrictions on the number of alcohol vendors should be used to control alcohol consumption.
- Advertising of alcohol either directly or indirectly should be banned.
- Primary care facilities for managing alcohol use disorders need to be made widely available.
Classification and natural history of alcoholic liver disease
Excessive alcohol consumption most often causes fat accumulation of hepatocytes, called hepatic steatosis (Figure 1). Alcohol-induced steatosis is in general reversible after alcohol abstinence. Continued alcohol overconsumption in the presence of steatosis markedly increases the risk for development of hepatitis, fibrosis and cirrhosis (Teli 1995, Cubero 2009). Patients with alcohol-induced cirrhosis have a significantly increased risk for hepatocellular carcinoma (McCollough 1998). Patients who only have fatty liver in the absence of inflammation and fibrosis have a much lower risk for development of cirrhosis than those with fatty liver plus presence of inflammation and fibrosis. The latter group of patients with alcoholic fatty liver, inflammation and fibrosis is defined as alcoholic steatohepatitis (ASH). The liver histology of patients with ASH is similar when compared to patients with non-alcoholic steatohepatitis (NASH) that is often associated with obesity and diabetes (Ludwig 1980, Brunt 1999).
The diagnosis of ASH by liver biopsy thus helps to define the risk for development of cirrhosis. The histological diagnosis of ASH however should not be confused with the term “alcoholic hepatitis” (also called “acute alcoholic hepatitis”) although its course can be a rather chronic one (Lucey 2009). This overview article concentrates on alcoholic hepatitis, which is a clinical diagnosis of a rather acute development of jaundice and liver failure associated with a high short-term mortality. In contrast to alcoholic hepatitis, there is as yet no specific pharmacological therapy for alcoholic cirrhosis (EASL 2012).
The factor(s) that set off the development of severe alcohol hepatitis are not exactly known. In general, pathogenesis and individual predisposition are governed by gene-environment interactions in all types of alcoholic liver disease (Figure 1). Based on the second-hit or multiple-hits hypothesis, patients are predisposed to progressive alcoholic liver disease when a specific combination of gene and environmental interaction exists (Tsukamoto 2009). A loss or gain of function genetic model has become a popular experimental approach to test the role of a gene as a second hit. Significant interactions for progressive development of alcoholic liver disease have been proven in particular for female gender, obesity, various drugs, iron overload, and hepatitis B and C viral infections (Mueller 2009, Machado 2009, Cubero 2009). These factors may also interact in the development of hepatocellular carcinoma (HCC).
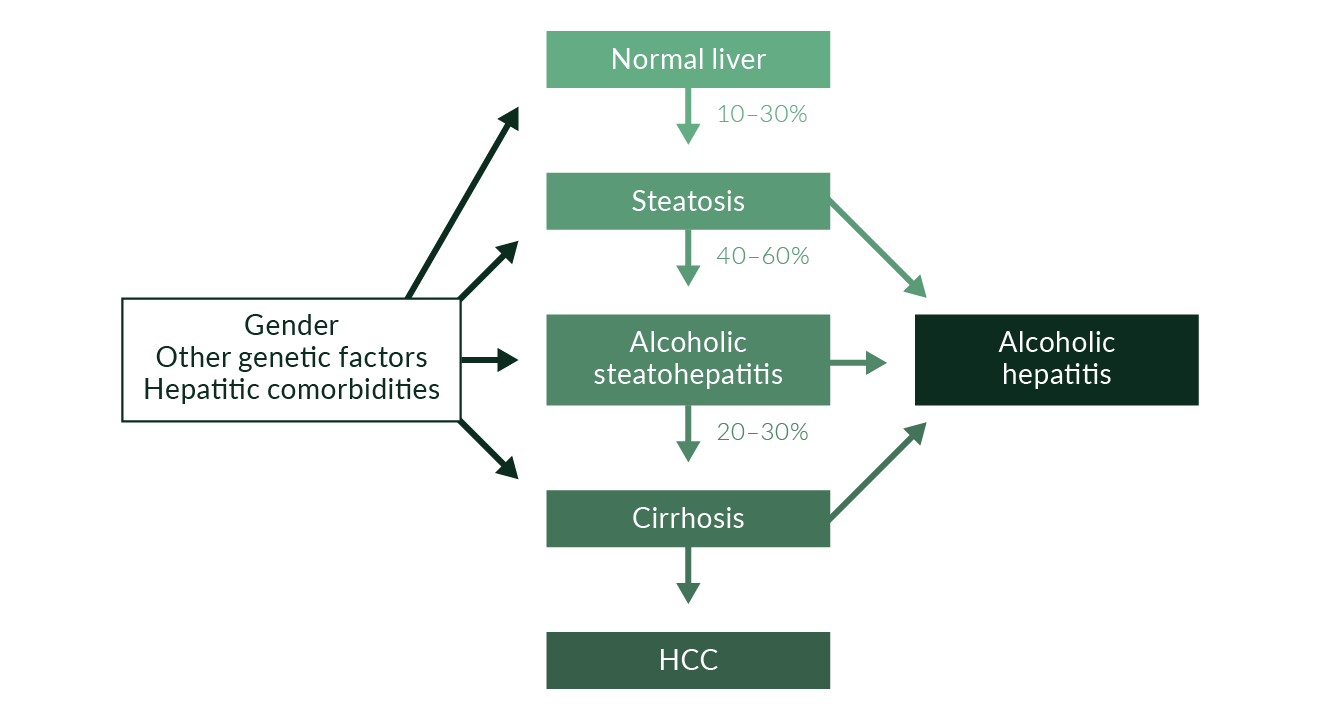 Figure 1. Effects of alcohol overconsumption on the liver
Figure 1. Effects of alcohol overconsumption on the liver
A liver biopsy in someone with alcoholic hepatitis is often similar to a histological feature of ASH. Most patients with histological features of ASH however will not develop alcoholic hepatitis. Alcohol overconsumption leads to a severe form of hepatitis and liver failure associated with a high short-term mortality only in some subjects. Such alcoholic hepatitis may be seen with or without preexisting cirrhosis.
Clinical features and diagnosis of alcoholic hepatitis
Alcoholic hepatitis is a clinical diagnosis characterised by the rapid development of jaundice and liver failure most often due to long-term alcohol overconsumption (Naveau 1997, McCollough 1998, Lucey 2009). Further characteristics include fever, ascites, and in some patients hepatic encephalopathy as well. Usually the liver is enlarged and tender. Women have a higher risk for alcoholic hepatitis than men assuming that both genders drink the same amount of alcohol. The type of alcohol is not associated with the risk. Prevalence was 20% in a cohort of 1604 patients who had a history of heavy alcohol consumption and underwent a liver biopsy (Naveau 1997).
Laboratory markers include increases in serum aspartate aminotransferase (AST) to approximately twice the upper limit of normal (ULN), while the increase in alanine aminotransferase (ALT) is less pronounced. The ratio of AST to ALT is typically >2 (Cohen 1979, Matloff 1980). Other laboratory abnormalities include increases in peripheral leukocytes, serum bilirubin and international normalised ratio (INR) (Mathurin 2002, Orrego 1979). In the presence of an increase in serum creatinine there is a high risk for development of an often lethal hepatorenal syndrome (Multimer 1993).
A liver biopsy usually shows large fat droplets and ballooning of hepatocytes that may also include alcoholic hyaline (also called Mallory bodies); these changes are accompanied by neutrophil infiltration and intrasinusoidal fibrosis (Figures 2 and 3) (MacSween 1986).
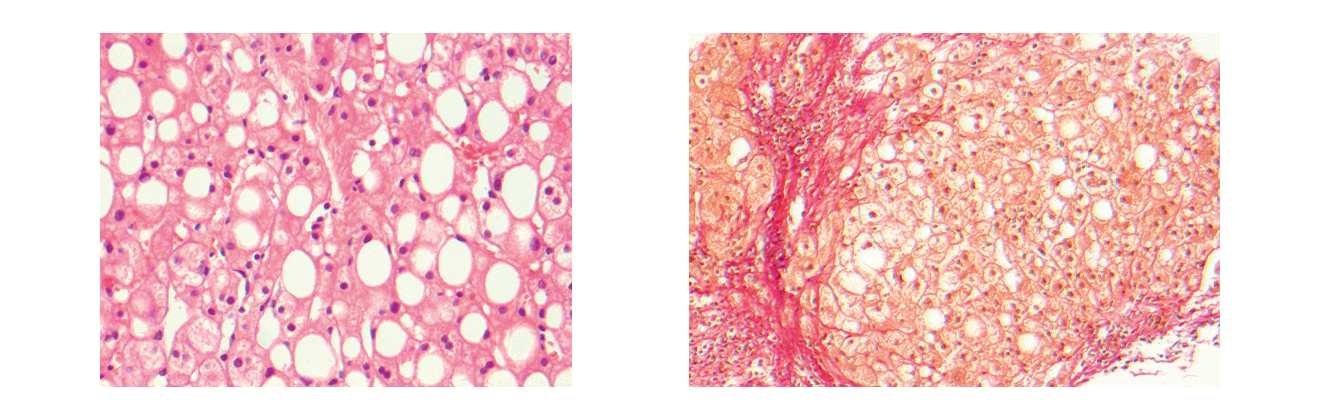 Figures 2 and 3. Liver biopsies of alcoholic hepatitis
Figures 2 and 3. Liver biopsies of alcoholic hepatitis
The diagnosis of alcoholic steatohepatitis (ASH) requires the presence of fibrosis. The role of liver biopsy in defining prognosis and treatment of alcoholic hepatitis in the clinical setting remains unclear. Currently, prognosis is usually based on clinical scoring systems and not on liver biopsy (Lucey 2009).
Ultrasound is routinely done to look for hepatocellular carcinoma, biliary obstruction, ascites, splenomegaly, portal vein thrombosis, and signs of portal hypertension. Ascites should be checked for spontaneous bacterial peritonitis routinely.
Differential diagnosis of alcoholic hepatitis includes severe non-alcoholic steatohepatitis (NASH), acute or chronic viral hepatitis, drug-induced injury, autoimmune hepatitis, and Wilson’s disease. NASH shares the histological features of ASH except for the rapid development of jaundice and liver failure.
After discontinuation of alcohol consumption the majority of patients will recover from alcoholic hepatitis, although jaundice, ascites and encephalopathy may persist for weeks or months (Alexander 1971). However, alcoholic hepatitis causes increased mortality in a considerable percentage of patients, despite adequate treatment and abstinence (Mathurin 2002, Orrego 1979).
Until recently, there was no histologic classification system to determine the prognosis of patients with alcoholic hepatitis. A recent study evaluated the histologic features associated with disease severity and proposed a histologic score to predict short-term (90-day) mortality (Altamirano 2014). The primary analysis included data from 121 patients admitted to a liver clinic in Barcelona, Spain. The system was updated in a test set of 96 patients from 5 academic centres in the United States and Europe, and a semiquantitative scoring system called the Alcoholic Hepatitis Histologic Score (AHHS) was developed (Alamirano 2014). The system was validated with an independent group of 109 patients. The degree of fibrosis and of neutrophil infiltration, type of bilirubinostasis, and presence of megamitochondria were independently associated with 90-day mortality. These four parameters are used for the AHHS to identify patients with a low (0–3 points), moderate (4–5 points), or high (6–9 points) risk of death within 90 days (3%, 19%, and 51%, respectively; p < 0.0001). The AHHS estimated 90-day mortality in the training and test sets with an area under the receiver operating characteristic analysis of 0.77 (95% confidence interval 0.71–0.83). The AHHS thus is likely to be useful in clinical decision-making.
Course and severity
Severe alcoholic hepatitis occurs in a small fraction of patients who have high alcohol consumption. In most cohorts, the 28-day mortality is high and ranges from 30% to 50% (Cohen 2009). Various scores have been used to predict the prognosis of alcoholic hepatitis. Maddrey’s discriminant function (Maddrey 1978) and the Model for End-Stage Liver Disease (MELD) score may help to identify patients who can benefit from corticosteroids. Most scores share some important characteristics such as serum bilirubin and prothrombin time (Srikureja 2005). Maddrey’s discriminant function is calculated using the equation: [4.6x (prothrombin time–control prothrombin time, in seconds)] + serum bilirubin (mg/dL). A value of >32 indicates severe alcoholic hepatitis and consequently calls for the use of corticosteroids (Maddrey 1978). In two retrospective studies, the MELD score predicted short-term mortality in alcoholic hepatitis as well as, or better than, Maddrey’s discriminant function (Dunn 2005, Srikureja 2005). A MELD score >21 was associated with a 90-day mortality of 20%. The Lille score is based on pretreatment data and on the response of serum bilirubin to a 7-day treatment with corticosteroids and has been used to determine whether corticosteroids should be discontinued after 7 days because of treatment failure (Forrest 2005, Dunn 2005, Louvet 2007). Patients with Maddrey’s discriminant function of <32 usually have mild disease with a short-term survival of more than 90% and will not benefit from corticosteroid treatment.
Investigators reported the results of a stepwise logistic-regression identifying variables related to survival 1–4 months after hospital admission in patients with alcoholic hepatitis (Forrest 2005); by using this data the Glasgow alcoholic hepatitis score was developed (not to be confused with the Glasgow coma score). The score, which includes age, peripheral leukocytes, urea nitrogen, bilirubin, and prothrombin time, may help to identify high-risk patients who should receive corticosteroids. Patients with a Maddrey’s discriminant function >32 and a Glasgow alcoholic hepatitis score of >9 who were treated with corticosteroids had an 84-day survival of 59%, while untreated patients had a 38% survival (Forrest 2007). In one study, the Glasgow score indicated which subgroup of patients with a high score of Maddrey’s discriminant function would benefit from corticosteroid therapy (Forrest 2007).
Child-Pugh (CP) and MELD scores have been widely used to predict survival in cirrhotic patients. Recent studies have suggested that the addition of serum sodium to MELD (MELD-Na score) may improve its prognostic accuracy. Another recent study compared the performance of CP, MELD, and MELD-Na scores in predicting 6-month mortality in patients with alcoholic cirrhosis, and developed a new prognostic score (Demy 2009). In this study, two French centres randomised 520 patients (mean age 56.4±10.2 years) with alcoholic cirrhosis into two groups. MELD, MELD-Na1, and MELD-Na2 were calculated according to UNOS recommendations. Frequencies of CP classes were: A – 29.6%, B – 25.8%, C – 44.6%. Of the 520 patients, 93 died during the 6-month follow-up. In the whole population, the values of CP, MELD, MELD-Na1, and MELD-Na2 for prediction of 6-month mortality were similar. Multivariate analysis identified age, bilirubin, urea, prothrombin time, sodium, and alkaline phosphatase as independent predictors of 6-month mortality. The score combining these six variables was named the Prognostic Score for Alcoholic Cirrhosis (PSAC) and compared to the four other scores. The predictive value of PSAC was better than all other scores except for MELD-Na2. By stepwise multivariate analysis, PSAC was identified as independently associated with 6-month mortality at the first step, and CP at the second. The new PSAC score may improve the prognostic accuracy to predict the 6-month outcome (Demy 2009).
Another study analysed the outcome of 79 patients who were admitted to an Intensive Care Unit (ICU) because of alcoholic liver disease (Rye 2009). The value of various scores was analysed for predicting mortality including the Acute Physiology, Age and Chronic Health Evaluation (APACHE II), Sequential Organ Failure Assessment (SOFA), CP, and MELD scores. The major reason for admission was sepsis (44%). The observed mortality in the ICU was 49% and hospital mortality 68%. Compared to survivors, non-survivors had a significantly higher serum bilirubin, creatinine and prothrombin time, and lower GCS and length of ICU stay. Survival was affected by cardiac arrest pre-admission (mortality 75%) and number of organs supported (mortality 8% with no organ support, 79% ≥ 2 organs, 100% ≥ 3 organs). Renal replacement therapy was associated with 100% mortality. Mortality due to GI bleeding was only 33%. Thus, cirrhotic patients who are admitted to ICU with cardiac arrest pre-admission, the need for renal replacement therapy, or multiple organ support, have a poor prognosis. The predictive accuracy of SOFA and MELD scores were superior to APACHE II and Child-Pugh scores in cirrhotic patients (Rye 2009).
A further study analysed the mortality of 105 patients presenting with alcoholic hepatitis (Hussain 2009). Patients were evaluated by the modified discriminant function (mDF) for alcoholic liver disease, CP score, and Glasgow alcoholic hepatitis score (GAHS). Mean survival for those alive at the end of the study (n=36) was 34.6 ± 17.8 months. Mean survival for those who died (n=50) was 13.2 ± 14.4 months. The mDF, CP and GAHS scores were significant predictors of mortality in this population. Prothrombin time was also a significant predictor of mortality (Hussain 2009).
Mechanisms of alcohol-related liver injury
Alcoholic liver disease is initiated by different cell types in the liver and a number of different factors including products derived from alcohol-induced inflammation, ethanol metabolites, and indirect reactions from those metabolites, as well as genetic predisposition (Colmenero 2007). Ethanol oxidation results in the production of metabolites that have been shown to bind and form protein adducts, and to increase inflammatory, fibrotic and cirrhotic responses. Lipopolysaccharide (LPS) has many deleterious effects and plays a significant role in a number of disease processes by increasing inflammatory cytokine release. In alcoholic liver disease, LPS is thought to come from a breakdown in the intestinal wall enabling LPS from resident gut bacterial cell walls to leak into the blood stream. The ability of adducts and LPS to independently stimulate various cells of the liver provides for a two-hit mechanism by which various biological responses are induced and result in liver injury.
Alcohol (ethanol) can be oxidised by various enzymatic and non-enzymatic pathways (Figure 4). In hepatocytes, the most important pathway is oxidation of ethanol via alcohol dehydrogenase (ADH) to acetaldehyde (Figure 4). In mitochondria, acetaldehyde is converted to acetate and in turn acetate is converted to acetyl CoA, which leads the two-carbon molecule into the TCA (tricarboxylic acid cycle).
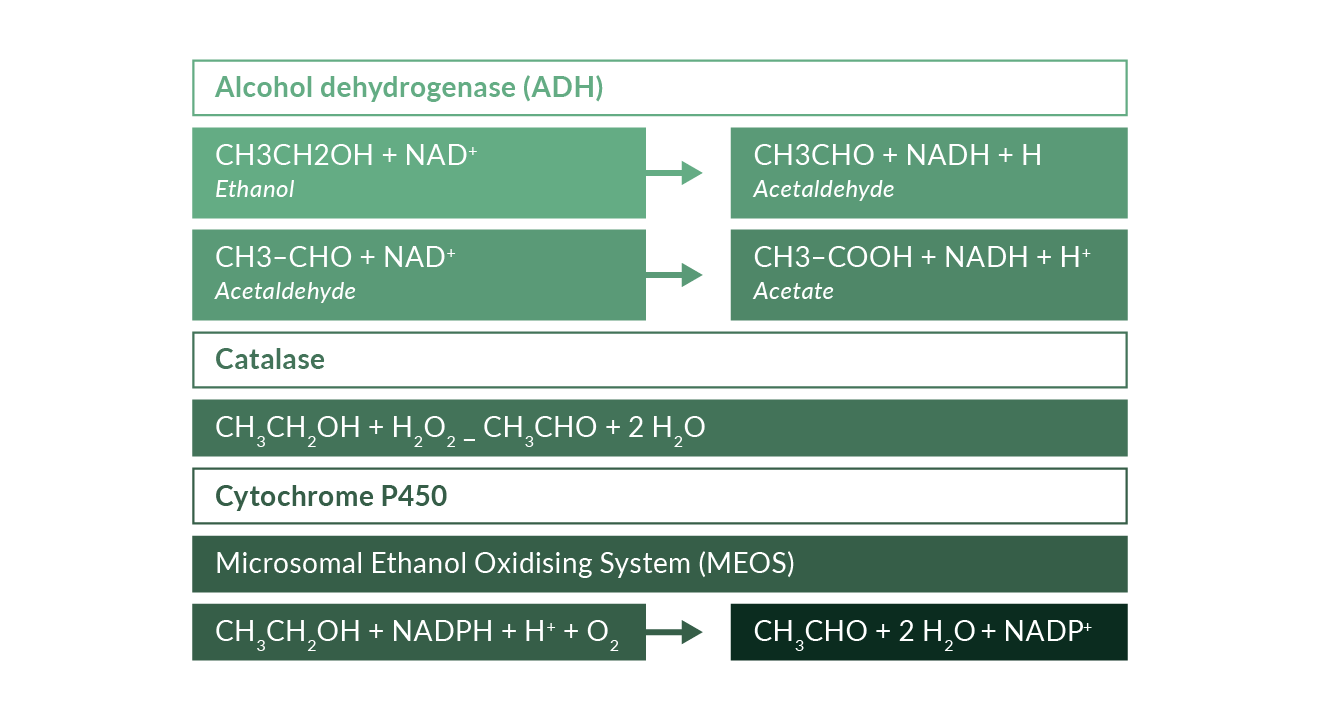 Figure 4. Oxidation of ethanol to acetaldehyde by enzymatic pathways
Figure 4. Oxidation of ethanol to acetaldehyde by enzymatic pathways
This oxidation generates reducing equivalents, primarily reduced nicotinamide adenine dinucleotide (NAD), i.e., NADH. The changes in the NADH–NAD+ potential in the liver inhibit both fatty acid oxidation and the TAC and may thereby increase lipogenesis (You 2004a). Ethanol has also been shown to increase lipid metabolism by inhibiting peroxisome-proliferator–activated receptor α (PPARα) and AMP kinase as well as by stimulation of sterol regulatory element-binding protein (Fischer 2003, You 2004b, Ji 2006). All these mechanisms lead to hepatic steatosis. Further enzymatic pathways of ethanol oxidation include catalase and the “Microsomal Ethanol Oxidising System” (MEOS), a cytochrome P450 component. Oxidation of ethanol to acetaldehyde may also be due to non-enzymatic free radical pathways (Figure 5). These pathways include strong oxidising intermediates such as the hydroxyl radical which can abstract a hydrogen atom from ethanol, preferentially producing the 1-hydroxyethyl radical; hypervalent iron complexes may also catalyse this reaction without involvement of •OH (Reinke 1994, Welch 2002, Qian 1999). Hydroxyethyl radicals may then react with oxygen to form a peroxy radical intermediate which can rearrange to release acetaldehyde and superoxide. Hydroxyethyl radicals can also react with proteins to produce antigenic adducts or induce mitochondrial permeability transition (Clot 1995, Sakurai 2000).
There are probably various other mechanisms by which ethanol may cause or contribute to liver disease. Ethanol increases the translocation of lipopolysaccharide (LPS) from the small and large intestines to the portal vein and on to the liver. In Kupffer cells, LPS can bind to CD14, which combines with toll-like receptor 4 (TLR4) thereby activating multiple cytokine genes (Schaffert 2009). In addition, NADPH oxidase may release reactive oxygen species (ROS) that activate cytokine genes within Kupffer cells, hepatocytes, and hepatic stellate cells. These cytokines including TNF-α may cause fever, anorexia, and weight loss. Interleukin-8 and monocyte chemotactic protein 1 (MCP-1) have been shown to attract neutrophils and macrophages. Platelet-derived growth factor (PDGF) and transforming growth factor b (TGF-b) contribute to the activation, migration, and multiplication of hepatic stellate cells, thereby promoting liver fibrosis.
 Figure 5. Oxidation of ethanol to acetaldehyde by non-enzymatic free radical pathways
Figure 5. Oxidation of ethanol to acetaldehyde by non-enzymatic free radical pathways
In the hepatocyte, ethanol is converted to acetaldehyde by the cytosolic enzyme alcohol dehydrogenase (ADH) and the microsomal enzyme cytochrome P450 2E1 (CYP2E1). Acetaldehyde is converted to acetate. These reactions produce NADH and inhibit the oxidation of triglycerides and fatty acids. ROS released by CYP2E1 and mitochondria cause lipid peroxidation. Inhibition of proteosomes due to ethanol disturbs protein catabolism and may be partly responsible for the formation of Mallory bodies. Reduction in enzymes that convert homocysteine to methionine may increase homocysteine, thereby injuring the endoplasmic reticulum. Decrease in binding of peroxisome proliferator–activated receptor alpha (PPAR-α) to DNA reduces the expression of genes involved in fatty acid oxidation.
Glutathione transport from the cytosol into the mitochondria is reduced by ethanol. Ethanol may also activate Fas and TNF receptor 1 (TNF-R1) thereby activating caspase 8, causing mitochondrial injury and opening the mitochondrial transition pore (MTP), releasing cytochrome c, and activating caspases; all these processes contribute to apoptosis. Activation of TNF-R1 leads to nuclear factor kappa B (NFkB) activation (Schaffert 2009).
Gut permeability and the circulating LPS endotoxin levels of the outer wall of gram negative bacteria are increased in patients with alcoholic liver injury (Uesugi 2002, Bjarnason 1984, Urbaschel 2001). In various animal studies, alcohol exposure promoted the transfer of LPS endotoxins from the intestine into portal blood (West 2005). Oral treatment with antibiotics reduced such increases in LPS endotoxins and ameliorated alcoholic liver injury in animals (Uesugi 2001, Nanji 1994, Adachi 1995). Activation of Kupffer cells by LPS endotoxins involves CD14, toll-like receptor 4 (TLR4), and MD2 (Uesugi 2001, Akira 2001, Yin 2001). The downstream pathways of TLR4 signalling include activation of early growth response 1 (EGR1), NFkB, and the TLR4 adapter also called toll-interleukin-1 receptor domain-containing adapter inducing interferon-ß (TRIF) (McCuillen 2005, Zhao 2008). TRIF-dependent signaling may contribute to alcohol-induced liver damage mediated by TLR4 (Hritz 2008).
Many animal studies have also shown that alcohol increases various markers of oxidative stress (Meagher 1999, Wu2009). Studies in rats and mice suggest that activated macrophages (Kupffer cells) and hepatocytes are the main sources of alcohol-induced free radicals (Bailey 1998, Kamimura 1992). Oxidative stress may mediate alcohol-induced liver injury, e.g., via cytochrome P450 2E1 (Mansuri 1999, Lu 2008), leading to mitochondrial damage, activation of endoplasmic reticulum–dependent apoptosis, and up-regulation of lipid synthesis (Ji 2003, Yin 2001). Activated Kupffer cells will also release TNF-α; this cytokine plays an important role in the pathogenesis of alcoholic hepatitis. Circulating TNF-α concentrations are higher in patients with alcoholic hepatitis than in heavy drinkers with inactive cirrhosis, heavy drinkers who do not have liver disease and persons who do not drink alcohol and who do not have liver disease (Adachi 1994, Bird 1990). Circulating TNF-α concentrations are associated with high mortality (Bird 1990). In animal studies, knockouts of the TNF receptor 1 and administration of the anti-TNF agent thalidomide both ameliorated alcohol-induced liver injury (Yin 1999, Imuro 1997, Enomoto 2002). Ethanol was also shown to release mitochondrial cytochrome c and to induce expression of the Fas ligand that may then cause apoptosis via the caspase-3 activation pathway (Zhou 2001). Both TNF- and Fas-mediated signals may increase the vulnerability of hepatocytes (Minagawa 2004).
Role of PNPLA3 polymorphisms and other genetic factors in the progression of alcoholic liver disease
The genetic determinants of the pathogenesis and disease progression of alcoholic and non-alcoholic fatty liver disease remained obscure until recently. In 2008, two genome-wide association (GWAS) studies linked the rs738409 polymorphism (I148M) of patatin-like phospholipase domain containing 3 (PNPLA3) with hepatic fat content and ALT levels (Romeo 2008, Yuan 2008). Later several further studies corroborated such association between the I148M polymorphism and NAFLD in almost all ethnic and age groups (Shang 2014, DiStefano 2014, Baclig 2014, Trepo 2014; for further literature see chapter on non-alcoholic fatty liver disease). The I148M polymorphism also predisposed to cirrhosis (Shen 2014) and hepatocellular carcinoma (Burza 2012, Valenti 2013, Trepo 2014). Other recent data suggest that the IL48M PNPLA3 polymorphism also accelerates fibrosis progression and HCC incidence in alcoholic liver disease (Trepo 2012, Nault 2014, Buch 2015, Stickel 2015, Falleti 2015). A recent genome-wide association study (GWAS) confirmed PNPLA3 and identified TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis (Buch 2015). All three loci are known to have a role in lipid processing, suggesting that lipid turnover is important in the pathogenesis of alcohol-related cirrhosis. Another recent study assessed the interaction between PNPLA3 rs738409 and TM6SF2 rs58542926 variants for HCC development (Falleti 2015). The results showed that TM6SF2 C/T or T/T in conjunction with PNPLA3 G/G variants may be potential genetic risk factors for developing HCC in alcohol-related cirrhosis (Falleti 2015). Details and mechanisms are discussed in a recent review about the genetics of alcoholic liver disease (Anstee 2015).
Treatment
Abstinence from alcohol
Continued alcohol use after diagnosis of alcoholic liver disease is the most important risk factor for complications and death (EASL 2012, Bell 2004). In such patients the development of new episodes of ASH are associated with a bad prognosis. Nicotin use has also been shown to be associated with mortality in patients with alcoholic liver disease (Pessione 2003). Other comorbid diseases further increase the risk of both cirrhosis-related and non-cirrhosis-related death (Jepsen 2008). Thus, after recovery from liver failure all patients with alcoholic hepatitis need to have psychological and social support to assure continued abstinence (Saitz 2007).
Supportive therapy
There is still a lack of specific therapy for patients with alcoholic hepatitis although prednisolone and pentoxifylline may have beneficial effects in severe disease. It is, however, generally accepted that all complications and risks such as ascites, encephalopathy, hepatorenal syndrome, and infections should be treated like other decompensated liver diseases (Kosten 2003, Sanyal 2008, Lim 2008). Daily protein intake should be at least 1.5 g/kg. Vitamin B1 and other vitamins should be administered according to recommended references (Barr 2006).
Corticosteroids
Various studies and meta-analyses show controversial results for the use of corticosteroids in alcoholic hepatitis (Imperiale 1990, Christensen 1999, Imperiale 1999, Rambaldi 2008). In general, corticosteroids have not been shown to increase survival, in particular during longer follow-up (Rambaldi 2008). However, there is evidence that corticosteroids do reduce mortality in a subgroup of patients with a Maddrey’s discriminant function >32 or in those presenting with hepatic encephalopathy (Rambaldi 2008). A meta-analysis of three studies corroborated that corticosteroids given for 28 days increase 1-month survival by 20% in severe alcoholic hepatitis (Maddrey’s discriminant function >32) (Mathurin 2002). In these studies Maddrey’s discriminant function >32 resembled a MELD score of >21. Prednisolone was usually given at 40 mg a day for 28 days. In some studies prednisolone was stopped completely at 28 days (Mathurin 2003), while the dose was gradually reduced in other studies (Imperiale 1990). Corticoids should not be given in the presence of sepsis, severe infection, hepatorenal syndrome, chronic hepatitis B, or gastrointestinal bleeding (O’Shea 2006).
The mechanisms by which corticosteroids improve short-term survival in severe alcoholic hepatitis are not fully understood. In general, corticosteroids inhibit various inflammatory processes by acting on activator protein 1 and NFkB (Barnes 1997). In patients with alcoholic hepatitis, some studies reported that corticosteroids were associated with a decrease in circulating levels of proinflammatory cytokines such as interleukin-8, TNF-α and others (Taieb 2000, Spahr 2001).
Recent reviews and recommendations conclude that corticosteroids should not be given to patients with a Maddrey’s discriminant function <32 or a MELD score <21 until further data can identify patients with a high short-term risk (Lucey 2009). Corticosteroids are thus ineffective in a large group of patients with alcoholic hepatitis and probably do not affect the long-term outcome. A randomised controlled clinical trial has shown that prednisolone 30 mg daily is superior to a broad antioxidant cocktail in the treatment of severe alcoholic hepatitis (Phillips 2006).
There is also evidence that corticosteroids can be discontinued after 7 days if there is no obvious improvement in clinical signs and symptoms and in serum bilirubin (Maddrey 1978, Dunn 2005, Forrest 2005, Louvet 2007).
The most recent Cochrane meta-analysis reported that corticosteroids significantly reduce mortality in trials that enrolled patients with a Maddrey score of at least 32 or with hepatic encephalopathy (Rambaldi 2008). Recent EASL and US guidelines thus also recommend corticosteroids for initial treatment of severe alcoholic hepatitis in the absence of sepsis and infections (O’Shea 2010, EASL 2012).
Pentoxifylline
In a randomised, controlled trial, pentoxifylline (400 mg TID for 28 days) reduced short-term mortality in severe alcoholic hepatitis (Maddrey’s discriminant function >32); mortality was 24% in the pentoxifylline group and 46% in the placebo group (p<0.01) (Akrivadis 2000). This study did not include a group on corticosteroid treatment. Although the phosphodiesterase inhibitor pentoxifylline has been suggested to act as an anti-TNF agent, TNF-α concentrations did not differ significantly between the two groups. Thus, the mechanisms by which pentoxifylline may improve the prognosis in alcoholic hepatitis remains unknown. Interestingly, almost all deaths (22 of 24; 92%) in the placebo group were associated with hepatorenal syndrome while hepatorenal syndrome was considered the cause of death in only 6 of 12 patients (50%) in the pentoxifylline group. Pentoxifylline might therefore exert its beneficial effects by preventing the development of hepatorenal syndrome. Another study (De BK 2009) compared the efficacy of pentoxifylline and prednisolone in the treatment of severe alcoholic hepatitis. This randomised double-blind controlled study enrolled 68 patients with severe alcoholic hepatitis (Maddrey score >32) who received either pentoxifylline (400 mg TID for 28 days) (n=34) or prednisolone (40 mg QD for 28 days) (n=34) for 28 days, with a subsequent open-label study (with a tapering dose of prednisolone) for a total of 3 months, and follow-up over 12 months. Twelve patients in the corticosteroid group died by the end of month 3 in contrast to five patients in the pentoxifylline group (mortality 35.3% vs 14.7%, p=0.04). Six patients in the corticosteroid group but none in the pentoxifylline group developed hepatorenal syndrome. Pentoxifylline was associated with a significantly lower MELD score at the end of 28 days of therapy when compared to corticosteroids (15.5 ± 3.6 vs 17.8 ± 4.6; p=0.04). Reduced mortality, improved risk: benefit profile and renoprotective effects of pentoxifylline compared with prednisolone suggest that pentoxifylline is superior to prednisolone for treatment of severe alcoholic hepatitis. Interestingly, long-term pentoxifylline therapy had also been shown to effectively achieve sustained biochemical improvement and even histological improvement in non-alcoholic steatohepatitis (Satapathy 2007). In a prospective, randomised study pentoxifylline (400 mg orally, three times a day for 4 weeks) for 4 weeks in patients with severe alcoholic hepatitis (Sidhu 2012) reduced mortality at 4 weeks compared to placebo (20% versus 40%; p=0.216). Renal failure was the cause of mortality in 20% of patients in the pentoxifylline group and in 70% of controls (p=0.11). Significant reduction in urea, creatinine, Maddrey score and TNF was noted in pentoxifylline group. This study showed that pentoxifylline improved renal and hepatic function with a trend towards decreased short-term mortality (Sidhu 2012). Pentoxifylline treatment is still recommended for severe alcoholic hepatitis by current EASL and US guidelines, in particular when the use of corticosteroids is risky for infection and sepsis (O’Shea 2010, EASL 2012).
Comparison and combination of corticosteroids and pentoxifylline
A multicentre, randomised, double-blind study evaluated whether the addition of pentoxifylline to prednisolone is more effective than prednisolone alone in patients with severe biopsy-proven alcoholic hepatitis (Mathurin 2013). The study included 270 French and Belgian heavy-drinkers with a recent onset of jaundice in the previous 3 months and a Maddrey score of at least 32. Patients were randomly assigned to receive either a combination of 40 mg of prednisolone once a day and 400 mg of pentoxifylline 3 times a day for 28 days, or 40 mg of prednisolone and matching placebo for 28 days. In an intention-to-treat analysis, 6-month survival was not different in the pentoxifylline-prednisolone vs. placebo-prednisolone groups (69.9% versus 69.2%; p = 0.91). By multivariate analysis, only the Lille model and the Model for End-Stage Liver Disease (MELD) scores were independently associated with 6-month survival. Also, the 7-day response and the incidence of hepatorenal syndrome at 6 months were not significantly different in the pentoxifylline-prednisolone and the placebo-prednisolone groups (Mathurin 2013).
Another study compared the efficacy of corticosteroids plus pentoxifylline with that of corticosteroids alone in patients with severe alcoholic hepatitis (Sidhu 2012). Four-week and six-month survival were not significantly different in the two groups (72.2% and 73.5%, respectively; p=1.00; 30.6% and 23.5%, respectively; p=0.417) (Sidhu 2012).
There is therefore no evidence that a combination of corticosteroids and pentoxifylline has an advantage compared with corticosteroids or pentoxifylline alone.
Although guidelines recommend the use of corticosteroids and/or pentoxifylline in severe alcoholic hepatitis (O’Shea 2010, EASL 2012), many studies reporting a benefit with these agents have methodological limitations. The STOPAH study, a multicentre, double-blind, factorial (2 × 2) trial, randomised 1,200 patients with severe alcoholic hepatitis in order to provide sufficient power to determine whether either of the two interventions is effective. Patients were randomised to one of four groups: Group A: placebo / placebo; Group B: placebo / prednisolone; Group C: pentoxifylline / placebo; Group D: pentoxifylline / prednisolone (Forrest 2013). The primary endpoint of the study is mortality at 28 days, with secondary endpoints being mortality at 90 days and 1 year (Forrest 2013). Preliminary results showed that steroids reduced mortality by 39% at day 28 without further sustained effects, whereas pentoxifylline did not have any beneficial effects. The final published results corroborated that pentoxifylline did not improve survival in patients with alcoholic hepatitis. Prednisolone was associated with a reduction in 28-day mortality that did not reach significance and with no improvement in outcomes at 90 days or 1 year. The authors conclude that pentoxifylline should not be used any longer for treatment of alcoholic hepatitis (Thurz 2015).
Recent reviews however still recommend to consider the use of pentoxifylline when prednisolone is contraindicated (Dugum 2015, Rahimi 2015, Liang 2015).
N-acetyl cysteine
A multicentre, randomised, controlled trial (Nguyen-Khac 2011) analysed treatment of severe acute alcoholic hepatitis via corticoids plus N-acetyl cysteine (C+NAC) versus corticoids (C) alone. The background to this approach was the hypothesis that the glutathione precursor NAC may rebuild antioxidant stocks in the hepatocyte. Deaths were significantly lower in the C+NAC group than in the C group at month 1 (n=7/85 (8.2%) vs. 21/89 (23.6%), p=0.005) and at month 2 (13/85 (15.3%) vs. 29/89 (32.6%), p=0.007) but not at month 3 (19/85 (22.4%) vs. 30/89 (33.7%), p=0.095) or at month 6 (23/85 (27.1%) vs. 34/89 (38.2%)). NAC may improve short-term survival. This improvement, however, is lost by month 3.
Anti-TNF-α therapy
Some smaller studies have shown beneficial results using the TNF-α receptor antagonists infliximab and etanercept in patients with acute alcoholic hepatitis (Spahr 2007, Mookerjee 2003, Tilg 2003, Menin 2004). A larger randomised, controlled clinical trial compared the effects of infliximab plus prednisolone vs placebo plus prednisolone in patients with severe alcoholic hepatitis (Maddrey’s discriminant function >32) (Naveau 2004). The trial was stopped early by the safety monitoring board because of a significant increase in severe infections and a (nonsignificant) increase in deaths in the infliximab group. Similarly, etanercept reduced 6-month survival when compared with placebo in a randomised, placebo-controlled trial (Boetticher 2008). Thus, TNF-α receptor antagonists should not be used for clinical therapy of alcoholic hepatitis (Lucey 2009).
Therapy with granulocyte colony-stimulating factor (G-CSF)
A recent randomised study evaluated the hypothesis that treating patients with alcoholic hepatitis with granulocyte colony-stimulating factor (G-CSF) might mobilise bone marrow-derived stem cells and promote hepatic regeneration and thereby improve survival (Singh 2014). One group received standard medical therapy and the other G-CSF at a dose of 5 μg/kg subcutaneously every 12 h for 5 consecutive days. There was a statistically significant increase in the number of CD34 (+) cells in the peripheral blood in the G-CSF group as compared with the standard therapy group after 5 days of therapy. There was also a significant reduction of survival and in Child-Pugh and MELD scores at 1, 2, and 3 months between the groups favouring G-CSF (Singh 2014). Further studies have to evaluate whether G-CSF is safe and effective in improving liver function and survival in patients with severe alcoholic hepatitis.
Nutritional support
Many patients with alcoholic hepatitis have signs of malnutrition associated with high mortality (Mendenhall 1984, Mendenhall 1986, Stickel 2003). Parenteral and enteral nutrition have been shown to improve malnutrition in alcoholic hepatitis but has not improved survival (Mendenhall 1984). A randomised, controlled clinical trial looked at the effects of enteral nutrition of 2000 kcal/day via tube feeding versus treatment with 40 mg/day prednisolone for 28 days in severe alcoholic hepatitis. Survival in both groups was similar after one month and one year. It may be concluded that nutritional support is as effective as corticosteroids in some patients (Cabre 2000). However, corticoids in many studies failed to improve long-term survival.
A randomised controlled trial comparing enteral nutrition versus corticosteroids did not show any difference in 28-day mortality rate (Cabre 2003). Indeed, deaths occurred earlier with enteral nutrition whereas steroid therapy was associated with a higher mortality rate in the weeks following the treatment period. Enteral nutrition probably deserves to be tested in combination with corticosteroids (EASL 2012). As yet, only one pilot study suggests that enteral nutrition associated with a short course of steroids may be a good therapeutic strategy for severe alcoholic hepatitis (Alvaraz 2004).
A recent randomised controlled trial determined whether the combination of corticosteroid and intensive enteral nutrition therapy is more effective than corticosteroid therapy alone in patients with severe AH (Moreno 2016). The study enrolled 136 heavy alcohol consumers with recent onset of jaundice and biopsy-proven severe AH; they were assigned randomly (1:1) to groups that received either intensive enteral nutrition plus methylprednisolone or conventional nutrition plus methylprednisolone (controls). In the intensive enteral nutrition group, enteral nutrition was given via feeding tube for 14 days. The primary end point was patient survival for 6 months. In the intention-to-treat analysis, there was no significant difference between groups in 6-month cumulative mortality: 44.4% in the enteral nutrition group vs. 52.1% in the controls (P = 0.406). Intensive enteral nutrition was difficult to implement and did not improve survival. However, further analysis showed that low daily energy intake was associated with greater mortality, so adequate nutritional intake should be a main goal for treatment.
Other pharmacologic treatments
The anabolic steroid oxandrolone failed to improve survival in patients with alcoholic hepatitis (Mendenhall 1984). Numerous studies have shown that alcoholic hepatitis is accompanied by oxidative stress. So far, all studies with antioxidants such as vitamin E, silymarin (milk thistle) and others have failed to improve survival in alcoholic hepatitis (Pares 1998, Mezey 2004). Older studies did show that colchicine, propylthiouracil, insulin and glucagon failed to improve survival in alcoholic hepatitis (Lucey 2009).
Liver transplantation
Alcoholic liver disease is still one of the most common indications for liver transplantation in Europe and in the US (Burra 2005, European Liver Transplant Registry 2011, Neuberger 1998, US Transplant Org 2011). In guidelines for liver transplantation, patients need to have at least a 6-month period of alcohol abstinence before they can be evaluated for transplantation, thus alcoholic hepatitis is usually a contraindication for liver transplantation (Lucey 1997, Everhardt 1997, Lucey 2007).
A substantial number of patients with severe alcoholic hepatitis fail to recover despite abstinence and medical therapy (Nakano 1982), and their chances for spontaneous recovery may be poor (Worner 1985). The classical opinion of European and North American experts considering acute alcoholic hepatitis as a contraindication for transplantation (EASL 2012) has recently been challenged by a case-control study showing an unequivocal improvement of survival in patients who received early transplantation (Mathurin 2011). Despite the fact that early liver transplantation for severe alcoholic hepatitis may improve survival in those patients who fail medical therapy, in many countries regulatory rules do not allow such transplants without documentation of six months of abstinence. Future evaluation of liver transplantation in carefully selected patients with severe alcoholic hepatitis who do not respond to standard medical therapy may be supported (Mathurin 2011).
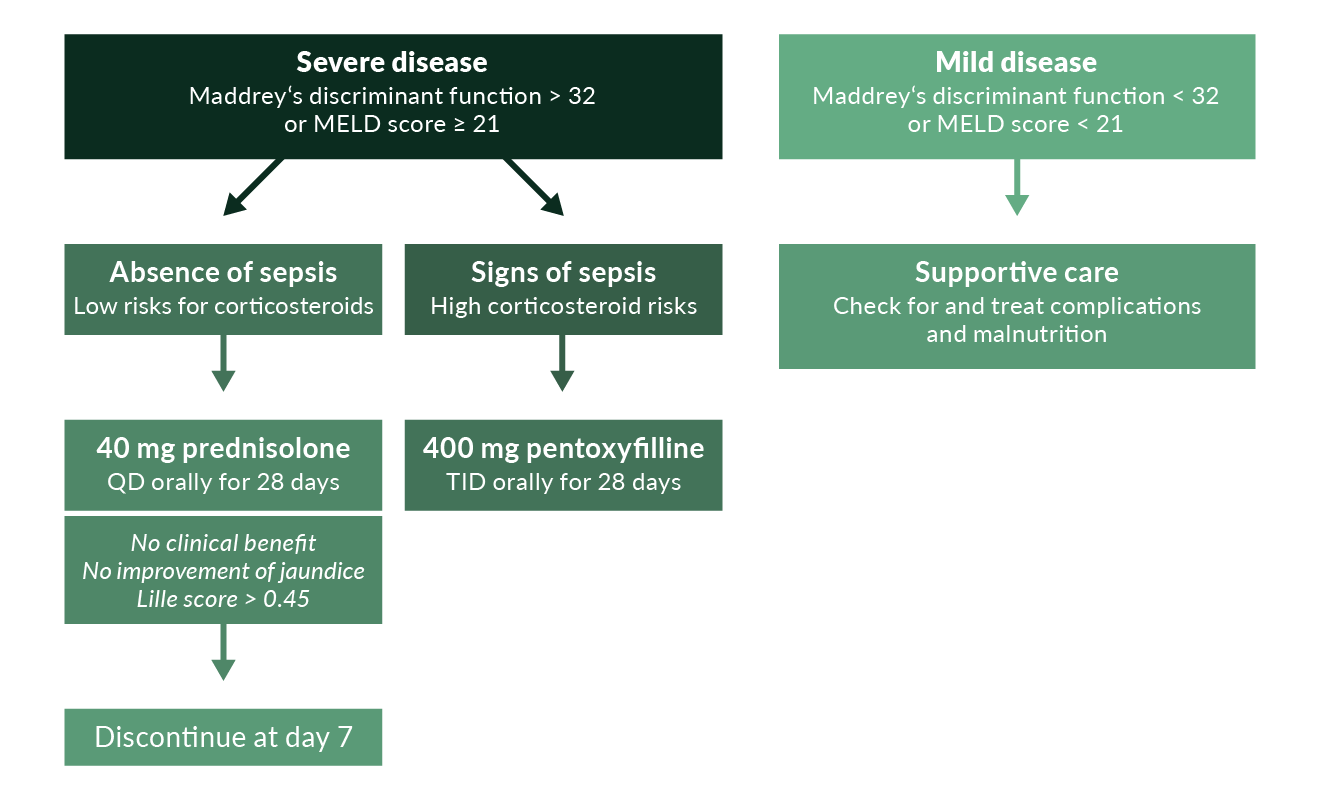 Figure 6. Treatment algorithm in alcoholic hepatitis. The use of pentoxifylline has recently been challenged by a large randomised trial (Thurz 2014); thus, its use is questionable
Figure 6. Treatment algorithm in alcoholic hepatitis. The use of pentoxifylline has recently been challenged by a large randomised trial (Thurz 2014); thus, its use is questionable
Summary
Alcoholic hepatitis is a clinical diagnosis based on a history of heavy alcohol consumption, jaundice, other signs of liver failure, and the absence of other causes of hepatitis. A liver biopsy may be helpful but is not required either to determine the diagnosis or prognosis. Abstinence from alcohol is the prerequisite for recovery. Patients with signs of malnutrition should have adequate nutritional support. Subjects with severe alcoholic hepatitis (Maddrey’s discriminant function >32 or MELD score >21) who do not have sepsis or other corticosteroid contraindications may receive 40 mg prednisolone daily for 28 days (McCullough 1998, Lucey 2009). A treatment algorithm based on current literature and EASL and US guidelines (O’Shea 2010, EASL 2012) is shown in Figure 6. After 7 days of corticosteroid treatment, patients without obvious clinical benefit, without significant improvement of jaundice and with a Lille score >0.45 may have disease that will not respond to continued treatment with corticosteroids or an early switch to pentoxifylline (Louvet 2008). In situations where administration of corticosteroids appears to be risky, pentoxifylline may be tried (Lucey 2009, O’Shea 2010, EASL 2012); this drug may decrease the risk of hepatorenal syndrome that is often lethal in alcoholic hepatitis. Patients with less severe alcoholic hepatitis have a good short-term survival of >90% and should not be treated with corticosteroids or pentoxyfilline (Mathurin 2002).
References
Adachi Y, Bradford BU, Gao W, et al. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 1994;20:453-460.
Adachi Y, Moore LE, Bradford BU, et al. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995;108:218-224.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001;2:675-680.
Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000;119:1637-1648.
Alexander JF, Lischner MW, Galambos JT, et al. Natural history of alcoholic hepatitis. II. The long-term prognosis. Am J Gastroenterol 1971;56:515-525.
Altamirano J, Miquel R, Katoonizadeh A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014;146:1231-9.
Anstee QM, Daly AK, Day CP. Genetics of Alcoholic Liver Disease. Semin Liver Dis 2015;35:361-74.
Bailey SM, Cunningham CC. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 1998;28:1318-1328.
Barnes PJ, Karin M. Nuclear factor-kappa B: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 1997;336:1066-1071.
Barr SI. Applications of Dietary Reference Intakes in dietary assessment and planning. Appl Physiol Nutr Metab 2006;31:66-73.
Becker U, Deis A, Sørensen TI, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology 1996;23:1025-1029.
Bell H, Jahnsen J, Kittang E, Raknerud N, Sandvik L. Long-term prognosis of patients with alcoholic liver cirrhosis: a 15-year follow-up study of 100 Norwegian patients admitted to one unit. Scand J Gastroenterol 2004;39:858–63.
Bellentani S, Saccoccio G, Costa G, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997;41:845-850.
Bird GL, Sheron N, Goka AK, et al. Increased plasma tumour necrosis factor in severe alcoholic hepatitis. Ann Intern Med 1990;112:917-920.
Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet 1984;1:179-182.
Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology 2008; 135:1953-1960.
Brunt EM, Janney CG, Di Bisceglie AM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467-2474.
Burra P, Lucey MR. Liver transplantation in alcoholic patients. Transpl Int 2005;18:491-498.
Cabré E, Rodríguez-Iglesias P, Caballería J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology 2000;32:36-42.
Christensen E, Gluud C. Glucocorticosteroids are not effective in alcoholic hepatitis. Am J Gastroenterol 1999;94:3065-3066.
Clot P, Bellomo G, Tabone M, et al. Detection of antibodies against proteins modified by hydroxyethyl free radicals in patients with alcoholic cirrhosis. Gastroenterology 1995;108:201-207.
Cohen JA, Kaplan MM. The SGOT/SGPT ratio - an indicator of alcoholic liver disease. Dig Dis Sci 1979;24:835-838.
Cohen SM, Ahn J. Review article: the diagnosis and management of alcoholic hepatitis.Aliment Pharmacol Ther 2009;30:3-13.
Colmenero J, Bataller R, Sancho-Bru P, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology 2007;132:687-697.
Cubero FJ, Urtasun R, Nieto N. Alcohol and liver fibrosis. Semin Liver Dis 2009;29:211-21.
De Alwis W, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis 2007;27:44-54.
De BK, Gangopadhyay S, Dutta D, et al. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol 2009;15:1613-9.
Demy M, Boursier J, Bertrais S, et al. Prediction of 6-months mortality in patients with alcoholic cirrhosis. Hepatology 2009;50:451A.
Dugum M, McCullough A. Diagnosis and Management of Alcoholic Liver Disease. J Clin Transl Hepatol 2015;3:109-16.
Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005;41:353-358.
EASL Clinical Practical Guidelines: Management of Alcoholic Liver Disease. J Hepatol 2012;57: 399-420. European Liver Transplant Registry. <http://www.eltr.org>; 2012.
Everhart JE, Beresford TP. Liver transplantation for alcoholic liver disease: a survey of transplantation programs in the United States. Liver Transpl Surg 1997;3:220-226.
Falleti E, Cussigh A, Cmet S, Fabris C, Toniutto P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig Liver Dis 2015. pii: S1590-8658(15)00622-2.
Fischer M, You M, Matsumoto M, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem 2003;278:27997-28004.
Forrest E, Mellor J, Stanton L, et al. Steroids or pentoxifylline for alcoholic hepatitis (STOPAH): study protocol for a randomised controlled trial. Trials 2013;14:262-8.
Forrest EH, Evans CD, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005;54:1174-1179.
Forrest EH, Morris AJ, Stewart S, et al. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut 2007;56:1743-1746.
Geall MG, Schoenfield LJ, Summerskill WHJ. Classification and treatment of chronic active liver disease. Gastroenterology 1968;55:724-9.
Gual A, Segura L, Contel M, Heather N, Colom J. AUDIT-3 and AUDIT-4:effectiveness of two short forms of the alcohol use disorders identification test. Alcohol Alcohol 2002;37:591-6.
Hritz I, Mandrekar P, Velayudham A, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008; 48:1224-1231.
Hussain S, Ali S, Moorthy D, et al. Predictors of Mortality in Alcoholic Liver Disease. Hepatology 2009;A273.
Imperiale TF, McCullough AJ. Do corticosteroids reduce mortality from alcoholic hepatitis? A meta-analysis of the randomized trials. Ann Intern Med 1990;13:299-307.
Imperiale TF, O’Connor JB, McCullough AJ. Corticosteroids are effective in patients with severe alcoholic hepatitis. Am J Gastroenterol 1999;94:3066-3068.
Jepsen P, Vilstrup H, Andersen PK, Lash TL, Sørensen HT. Co-morbidity and survival of Danish cirrhosis patients: a nationwide population-based cohort study. Hepatology 2008;48:214-20.
Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003;124:1488-1499.
Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol 2006;45:717-724.
Kamimura S, Gaal K, Britton RS, et al. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: association of lipid peroxidation with liver fibrogenesis. Hepatology 1992;16:448-453.
Kaner EF, Dickinson HO, Beyer F, et al. The effectiveness of brief alcohol interventions in primary care settings: a systematic review. Drug Alcohol Rev 2009;28:301-23.
Kosten TR, O’Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003;348:1786-1795.
Liang R, Liu A, Perumpail RB, et al. Advances in alcoholic liver disease: An update on alcoholic hepatitis. World J Gastroenterol 2015;21:11893-903.
Lim JK, Groszmann RJ. Vasoconstrictor therapy for the hepatorenal syndrome. Gastroenterology 2008;134:1608-1611.
Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol 2008;48:465-470.
Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007;45:1348-1354.
Lu Y, Zhuge J, Wang X, et al. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008;47:1483-1494.
Lucey MR, Brown KA, Everson GT, et al. Minimal criteria for placement of adults on the liver transplant waiting list: a report of a national conference organized by the American Society of Transplant Physicians and the American Association for the Study of Liver Diseases. Liver Transpl Surg 1997;3:628-637.
Lucey MR. Liver transplantation for alcoholic liver disease: past, present, and future. Liver Transpl 2007;13:190-192.
Lucey MR, Connor JT, Boyer T, et al. Alcohol consumption by cirrhotic subjects: patterns of use and effects on liver function. Am J Gastroenterol 2008;103:1698-1706.
Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009;360:2758-69.
Ludwig J, Viggiano TR, McGill DB, et al. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980;55:434-438.
Machado MV, Ravasco P, Martins A, et al. Iron homeostasis and H63D mutations in alcoholics with and without liver disease. World J Gastroenterol 2009;15:106-11.
MacSween RN, Burt AD. Histologic spectrum of alcoholic liver disease. Semin Liver Dis 1986;6:221-232.
Maddrey WC, Boitnott JK, Bedine MS. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978;75:193-199.
Mathurin P, Moreno C, Samuel D, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med 2011;365:1790-1800.
Mathurin P, Louvet A, Duhamel A, et al. Prednisolone with vs without pentoxifylline and survival of patients with severe alcoholic hepatitis: a randomized clinical trial. JAMA 2013;310:1033-41.
Mathurin P, Abdelnour M, Ramond MJ, et al. Early change in bilirubin levels is an important prognostic factor in severe alcoholic hepatitis treated with prednisolone. Hepatology 2003;38:1363-1369.
Mathurin P, Mendenhall CL, Carithers RL Jr, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis (AH): individual data analysis of the last three randomized placebo controlled double blind trials of corticosteroids in severe AH. J Hepatol 2002;36:480-487.
Matloff DS, Selinger MJ, Kaplan MM. Hepatic transaminase activity in alcoholic liver disease. Gastroenterology 1980;78:1389-1392.
McCullough AJ, O’Connor JF. Alcoholic liver disease: proposed recommendations for the American College of Gastroenterology. Am J Gastroenterol 1998;93: 2022-2036.
Meagher EA, Barry OP, Burke A, et al. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest 1999;104:805-813.
Mendenhall CL, Anderson S, Weesner RE, et al. Protein-calorie malnutrition associated with alcoholic hepatitis. Am J Med 1984;76:211-222.
Mendenhall CL, Tosch T, Weesner RE, et al. VA cooperative study on alcoholic hepatitis. II: Prognostic significance of protein-calorie malnutrition. Am J Clin Nutr 1986;43:213-218.
Mendenhall CL, Anderson S, Garcia-Pont P, et al. Short-term and long-term survival in patients with alcoholic hepatitis treated with oxandrolone and prednisolone. N Engl J Med 1984;311:1464-1470.
Mezey E, Potter JJ, Rennie-Tankersley L, et al. A randomized placebo controlled trial of vitamin E for alcoholic hepatitis. J Hepatol 2004;40:40-46.
Minagawa M, Deng Q, Liu ZX, et al. Activated natural killer T cells induce liver injury by Fas and tumour necrosis factor-alpha during alcohol consumption. Gastroenterology 2004;126:1387-1399.
Mokdad AH, Marks JS, Stroup DF, et al. Actual causes of death in the United States 2000. JAMA 2000;291:1238-1245.
Mookerjee RP, Sen S, Davies NA. Tumour necrosis factor alpha is an important mediator of portal and systemic haemodynamic derangements in alcoholic hepatitis. Gut 2003;52:1182-1187.
Moreno C, Deltenre P, Senterre C, et al. Intensive enteral nutrition Is ineffective for patients with severe alcoholic hepatitis treated with corticosteroids. Gastroenterology 2016;150:903-10.
Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol 2009;15:3462-71.
Mutimer DJ, Burra P, Neuberger JM, et al. Managing severe alcoholic hepatitis complicated by renal failure. Q J Med 1993;86:649-656.
Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease). Proc Soc Exp Biol Med 1994;205:243-247.
Nault JC, Nahon P. Genetic predisposition to hepatocellular carcinoma in alcoholic cirrhosis: The NCAN-PNPLA3-lipid connection? J Hepatol 2014;61:971-2.
Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology 1997;25:108-111.
Naveau S, Chollet-Martin S, Dharancy S, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004;39:1390-1397.
Neuberger J. Transplantation for alcoholic liver disease: a perspective from Europe. Liver Transpl Surg 1998;4:S51-S57.
Nguyen-Khac E, Thevenot T, Piquet M, et al. Treatment of severe Acute Alcoholic Hepatitis (AAH) with Corticoids plus N-Acetyl Cysteine versus Corticoids alone: a multicentre, randomized, controlled trial. N Engl J Med 2011;365(19):1781-9
Nishiguchi S, Kuroki T, Yabusako T, et al. Detection of hepatitis C virus antibodies and hepatitis C virus RNA in patients with alcoholic liver disease. Hepatology 1991;14:985-989.
Orrego H, Blake JE, Blendis LM, et al. Reliability of assessment of alcohol intake based on personal interviews in a liver clinic. Lancet 1979;2:1354-1356.
O’Shea R, McCullough AJ. Steroids or cocktails for alcoholic hepatitis. J Hepatol 2006; 44:633-636.
O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010;51:307-28.
Parés A, Planas R, Torres M, et al. Effects of silymarin in alcoholic patients with cirrhosis of the liver: results of a controlled, double-blind, randomized and multicenter trial. J Hepatol 1998;28:615-621.
Pessione F, Ramond MJ, Peters L, et al. Five-year survival predictive factors in patients with excessive alcohol intake and cirrhosis. Effect of alcoholic hepatitis, smoking and abstinence. Liver Int 2003;23:45-53.
Phillips M, Curtis H, Portmann B, et al. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis – a randomised clinical trial. J Hepatol 2006;44:784-90.
Qian SY, Buettner GR. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: an electron paramagnetic resonance spin trapping study. Free Radic Biol Med 1999;26:1447-1456.
Rahimi E, Pan JJ. Prognostic models for alcoholic hepatitis. Biomark Res 2015;3:20. eCollection 2015.
Rambaldi A, Saconato HH, Christensen E, et al. Systematic review: glucocorticosteroids for alcoholic hepatitis – a Cochrane Hepato-Biliary Group systematic review with meta-analyses and trial sequential analyses of randomized clinical trials. Aliment Pharmacol Ther 2008;27:1167-78.
Reinke LA, Rau JM, McCay M. Characteristics of an oxidant formed during iron (II) autoxidation. Free Rad Biol Med 1994;16:485-492.
Rye K, Krishnamoorthy R, Skene H, et al. Short - term prognosis and outcome predictors of alcohol - related cirrhosis admitted to the Intensive Care Unit. Hepatology 2009;50:427A.
Saitz R, Palfai TP, Cheng DM, et al. Brief intervention for medical inpatients with unhealthy alcohol use: a randomized, controlled trial. Ann Intern Med 2007;146:167-176.
Sakurai K, Stoyanovsky DA, Fujimoto Y, et al. Mitochondrial permeability transition induced by 1-hydroxyethyl radical. Free Rad Biol Med 2000;28:273-280.
Sanyal AJ, Boyer T, Garcia-Tsao G, et al. A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology 2008:134:1360-1368.
Satapathy SK, Sakhuja P, Malhotra V, et al. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol 2007;22:634-8.
Schaffert CS, Duryee MJ, Hunter CD, et al. Alcohol metabolites and lipopolysaccharide: roles in the development and/or progression of alcoholic liver disease. World J Gastroenterol 2009;15:1209-18.
Sidhu SS, Goyal O, Singla M, et al. Pentoxifylline in severe alcoholic hepatitis: a prospective, randomised trial. J Assoc Physicians India 2012;60:20-2.
Sidhu SS, Goyal O, Singla P, et al. Corticosteroid plus pentoxifylline is not better than corticosteroid alone for improving survival in severe alcoholic hepatitis (COPE trial). Dig Dis Sci 2012;57:1664-71.
Singh V, Sharma AK, Narasimhan RL, et al. Granulocyte colony-stimulating factor in severe alcoholic hepatitis: a randomized pilot study. Am J Gastroenterol 2014;109:1417-23.
Spahr L, Rubbia-Brandt L, Pugin J, et al. Rapid changes in alcoholic hepatitis histology under steroids: correlation with soluble intercellular adhesion molecule-1 in hepatic venous blood. J Hepatol 2001;35:582-589.
Spahr L, Rubbia-Brandt L, Frossard JL, et al. Combination of steroids with infliximab or placebo in severe alcoholic hepatitis: a randomized controlled pilot study. J Hepatol 2002;37:448-455.
Srikureja W, Kyulo NL, Runyon BA, et al. MELD score is a better prognostic model than Child-Turcotte-Pugh score or Discriminant Function score in patients with alcoholic hepatitis. J Hepatol 2005;42:700-706.
Stickel F, Hampe J, Trépo E, Datz C, Romeo S. PNPLA3 genetic variation in alcoholic steatosis and liver disease progression. Hepatobiliary Surg Nutr 2015;4:152-60.
Stickel F, Hoehn B, Schuppan D, et al. Nutritional therapy in alcoholic liver disease. Aliment Pharmacol Ther 2003;18:357-373.
Taïeb J, Mathurin P, Elbim C, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol 2000;32:579-586.
Teli MR, Day CP, Burt AD, et al. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995;346:987-990.
Thursz MR, Forrest EH, Ryder S; STOPAH investigators. Prednisolone or Pentoxifylline for Alcoholic Hepatitis. N Engl J Med 2015;373:282-3.
Tilg H, Jalan R, Kaser A, et al. Anti-tumour necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol 2003;38:419-425.
Trepo E, Guyot E, Ganne-Carrie N, et al. PNPLA3 (rs738409 C > G) is a common risk variant associated with hepatocellular carcinoma in alcoholic cirrhosis. Hepatology 2012;55:1307-8.
Trepo E, Nahon P, Bontempi G, et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014;59:2170-7.
Tsukamoto H, Machida K, Dynnyk A, et al. „Second hit” models of alcoholic liver disease. Semin Liver Dis 2009;29:178-87.
Uesugi T, Froh M, Arteel GE, et al. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001;34:101-108.
Uesugi T, Froh M, Arteel GE, et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 2002;168:2963-2969.
University of Sheffield Guidance Title: Prevention and early identification of alcohol use disorders in adults and young people. Final draft of Report 2 to the National Institute for Health & Clinical Excellence. Sheffield: The University of Sheffield, School of Health and Related Research (ScHARR); 2009.
Urbaschek R, McCuskey RS, Rudi V, et al. Endotoxin, endotoxin-neutralizing-capacity, sCD14, sICAM-1, and cytokines in patients with various degrees of alcoholic liver disease. Alcohol Clin Exp Res 2001;25:261-268.
US Transplant.org. <http://www.ustransplant.org/default.aspx>; 2013.
Vaillant GE. The natural history of alcoholism revisited. Boston: Harvard University Press 1995.
Valenti L, Al-Serri A, Daly AK, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209-17.
Welch KD, Davis Z, Aust SD. Iron autoxidation and free radical generation: effects of buffers, ligands, and chelators. Arch Biochem Biophys 2002;397:360-369.
Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis 2009; 29:141-54.
Yin M, Wheeler MD, Kono H, et al. Essential role of tumour necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999;117:942-952.
Yin M, Bradford BU, Wheeler MD, et al. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 2001;166:4737-4742.
Yin M, Gäbele E, Wheeler MD, et al. Alcohol-induced free radicals in mice: direct toxicants or signaling molecules? Hepatology 2001;34:935-942.
You M, Crabb DW, et al. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol 2004a; 287:G1-6.
You M, Matsumoto M, Pacold CM, et al. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004b;127:1798-1808.
Yuan X, Waterworth D, Perry JR, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 2008;83:520-8.
Zhao XJ, Dong Q, Bindas J, et al. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 2008;181:3049-3056.
Zhou Z, Sun X, Kang YJ. Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol 2001;159:329-338.