
24. Autoimmune liver diseases: AIH, PBC and PSC
Christian P. Strassburg
Autoimmune hepatitis (AIH)
Autoimmune hepatitis (AIH) is a chronic inflammatory disease, in which a loss of tolerance against hepatic tissue is presumed. Autoimmune hepatitis (AIH) was first described as a form of chronic hepatitis in young women showing jaundice, elevated gamma globulins and amenorrhoea, which eventually led to liver cirrhosis (Waldenström 1950). A beneficial effect of steroids was described in the reported patient cohort and thus the groundwork was laid for the first chronic liver disease found to be curable by drug therapy. AIH was later recognised in combination with other extrahepatic autoimmune syndromes, and the presence of antinuclear antibodies (ANA) led to the term lupoid hepatitis (Mackay 1956). Systematic evaluations of the cellular and molecular immunopathology, of the clinical symptoms and of laboratory features has subsequently led to the establishment of autoimmune hepatitis as a clinical entity on its own, which is serologically heterogeneous, treated by an immunosuppressive therapeutic strategy (Strassburg 2000). An established (Alvarez 1999a) and recently simplified (Hennes 2008b) revised scoring system allows for a reproducible and standardised approach to diagnosing AIH in a scientific context but has limitations in everyday diagnostic applications. The use and interpretation of seroimmunological and molecular biological tests permits a precise discrimination of autoimmune hepatitis from other etiologies of chronic hepatitis, in particular from chronic viral infection as the most common cause of chronic hepatitis worldwide (Strassburg 2002). Today, AIH is a treatable chronic liver disease in the majority of cases. Much of the same initial treatment strategies of immunosuppression still represent the standard of care. The largest challenge regarding treatment is the timely establishment of the correct diagnosis.
Definition and diagnosis of autoimmune hepatitis
In 1992, an international panel met in Brighton, UK, to establish diagnostic criteria for AIH because it was recognised that several features including histological changes and clinical presentation are also prevalent in other chronic liver disorders (Johnson 1993). In this and in a revised report the group noted that there is no single test for the diagnosis of AIH. In contrast, a set of diagnostic criteria was suggested in the form of a scoring system designed to classify patients as having probable or definite AIH (Table 1). According to this approach the diagnosis relies on a combination of indicative features of AIH and the exclusion of other causes of chronic liver diseases. AIH predominantly affects women of any age, and is characterised by a marked elevation of serum globulins, in particular gamma globulins, and circulating autoantibodies. It should be noted that AIH regularly affects individuals older than 40 but should be considered in all age groups (Strassburg 2006). The clinical appearance ranges from an absence of symptoms to a severe or fulminant presentation (Stravitz 2011) and responds to immunosuppressive treatment in most cases. An association with extrahepatic autoimmune diseases such as rheumatoid arthritis, autoimmune thyroiditis, ulcerative colitis and diabetes mellitus and a family history of autoimmune or allergic disorders has been reported (Strassburg 1995).
Autoantibodies are one of the distinguishing features of AIH. The discovery of autoantibodies directed against different cellular targets including endoplasmatic reticulum membrane proteins, nuclear antigens and cytosolic antigens has led to a suggested subclassification of AIH based upon the presence of three specific autoantibody profiles. According to this approach, AIH type 1 is characterised by the presence of antinuclear antibodies (ANA) and/or anti-smooth muscle antibodies (SMA) directed predominantly against smooth muscle actin. AIH type 2 is characterised by anti-liver/kidney microsomal autoantibodies (LKM-1) directed against cytochrome P450 CYP2D6 (Manns 1989, Manns 1991) (Figure 1) and with lower frequency against UDP-glucuronosyltransferases (UGT) (Strassburg 1996). AIH type 3 (Manns 1987, Stechemesser 1993) is characterised by autoantibodies against a soluble liver antigen (SLA/LP) identified as UGA suppressor serine tRNA-protein complex (Gelpi 1992, Wies 2000, Volkmann 2001, Volkmann 2010). However, this serological heterogeneity does not influence the decision of whom to treat or of what strategy to employ.
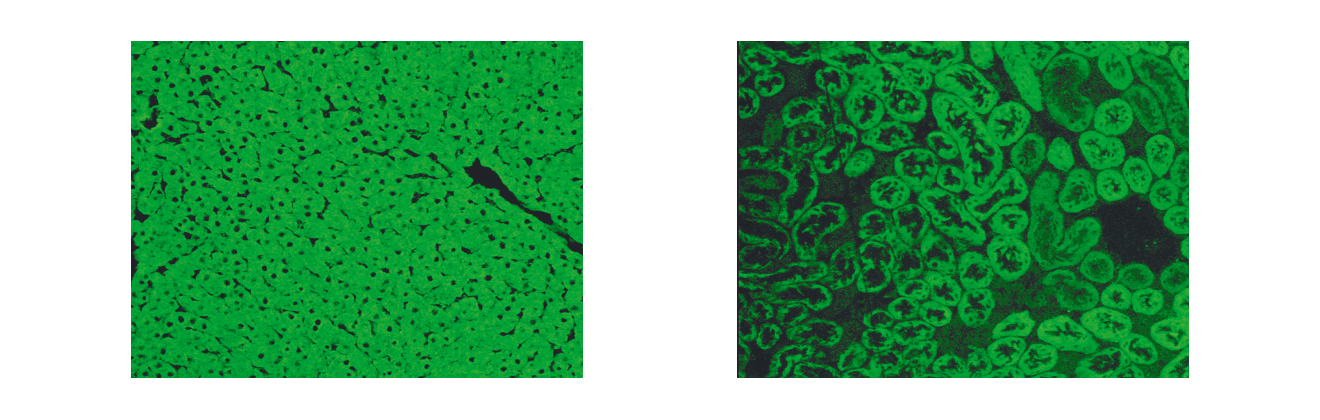 Figure 1. Indirect immunofluorescence showing LKM-1 autoantibodies on rat kidney and liver cryostat sections. Serum of a patient with autoimmune hepatitis type 2. A) Using rat hepatic cryostat sections a homogeneous cellular immunofluorescence staining is visualised excluding the hepatocellular nuclei (LKM-1). B) Typical indirect immunofluorescence pattern of LKM-1 autoantibodies detecting the proximal (cortical) renal tubules but excluding the distal tubules located in the renal medulla, which corresponds to the tissue expression pattern of the autoantigen CYP2D6
Figure 1. Indirect immunofluorescence showing LKM-1 autoantibodies on rat kidney and liver cryostat sections. Serum of a patient with autoimmune hepatitis type 2. A) Using rat hepatic cryostat sections a homogeneous cellular immunofluorescence staining is visualised excluding the hepatocellular nuclei (LKM-1). B) Typical indirect immunofluorescence pattern of LKM-1 autoantibodies detecting the proximal (cortical) renal tubules but excluding the distal tubules located in the renal medulla, which corresponds to the tissue expression pattern of the autoantigen CYP2D6
Although the histological appearance of AIH is characteristic, there is no specific histological feature that can be used to prove the diagnosis (Dienes 1989). Percutaneous liver biopsy is recommended initially for grading and staging (EASL 2015), as well as for therapeutic monitoring when this is considered necessary for therapeutic planning. Histological features usually include periportal hepatitis with lymphocytic infiltrates, plasma cells, and piecemeal necrosis. With advancing disease, bridging necrosis, panlobular and multilobular necrosis may occur and ultimately lead to cirrhosis. A lobular hepatitis can be present, but is only indicative of AIH in the absence of copper deposits or biliary inflammation. However, biliary involvement does not rule out AIH. The presence of granulomas and iron deposits argue against AIH.
Viral hepatitis should be excluded by the use of reliable, commercially available tests. Hepatitis E is frequently found in AIH patients and should be considered (van Gerven 2016). The exclusion of other hepatotropic viruses such as cytomegalovirus, Epstein-Barr and herpes may only be required in cases suspicious of such infections or if the diagnosis of AIH based on the above-mentioned criteria remains inconclusive.
The probability of AIH usually decreases whenever signs of bile duct involvement are present, such as elevation of alkaline phosphatase, histological signs of cholangiopathy and detection of AMA. If one or more components of the scoring system are not evaluated, only a probable diagnosis can be made (Table 1).
Epidemiology and clinical presentation
Based on limited epidemiological data, the prevalence is estimated to range between 20 to 50 cases per million among the Caucasian population in Western Europe and North America (Jepsen 2015). The prevalence of AIH is similar to that of systemic lupus erythematosus, primary biliary cholangitis and myasthenia gravis, which also have an autoimmune aetiology (Nishioka 1997, Nishioka 1998). Among the Caucasian population in North American and Western European, AIH accounts for up to 20% of cases with chronic hepatitis (Cancado 2000). However, chronic viral hepatitis remains the major cause of chronic hepatitis in most Western societies.
Autoimmune hepatitis is part of the syndrome of chronic hepatitis, which is characterised by sustained hepatocellular inflammation for at least six months and an elevation of ALT and AST of 1.5 times the upper limit of normal. In about 49% of AIH patients an acute onset of AIH is observed and rare cases of fulminant AIH have been reported. In most cases, however, the clinical presentation is not spectacular and is characterised by fatigue, right upper quadrant pain, jaundice and occasionally also by palmar erythema and spider naevi. In later stages, the consequences of portal hypertension dominate, including ascites, bleeding oesophageal varices and encephalopathy. A specific feature of AIH is the association of extrahepatic immune-mediated syndromes including autoimmune thyroiditis, vitiligo, alopecia, nail dystrophy, ulcerative colitis, rheumatoid arthritis, and also diabetes mellitus and glomerulonephritis.
Table 1. International criteria for the diagnosis of AIH (Alvarez 1999)| Parameter | Score |
| Gender Female Male | + 2 0 |
| Serum biochemistry Ratio of elevation of serum alkaline phosphatase to aminotransferase >3.0 1.5–3 <1.5 | – 2 0 + 2 |
| Total serum globulin, γ-globulin or IgG (x upper limit of normal) >2.0 1.5–2.0 1.0–1.5 <1.0 | + 3 + 2 + 1 0 |
| Autoantibodies (titres by immunfluorescence on rodent tissues) Adults ANA, SMA or LKM-1 >1:80 1:80 1:40 <1:40 | + 3 + 2 + 1 0 |
| Antimitochondrial antibody Positive Negative | – 4 0 |
| Hepatitis viral markers Negative Positive | + 3 – 3 |
| History of drug use Yes No | – 4 + 1 |
| Alcohol (average consumption) <25 gm/day >60 gm/day | + 2 - 2 |
| Genetic factors: HLA-DR3 or -DR4 | + 1 |
| Other autoimmune diseases | + 2 |
| Response to therapy Complete Relapse | + 2 + 3 |
| Liver histology Interface hepatitis Predominant lymphoplasmacytic infiltrate Rosetting of liver cells None of the above Biliary changes Other changes | + 3 + 1 + 1 – 5 – 3 – 3 |
| Seropositivity for other defined autoantibodies | + 2 |
Natural history and prognosis
Data describing the natural history of AIH are scarce. The last placebo-controlled immunosuppressive treatment trial containing an untreated arm was published in 1980 (Kirk 1980). The value of these studies is limited considering that these patients were only screened for then available epidemiological risk factors for viral hepatitis and were not characterised by standardised diagnostic criteria and available virological tests. Nevertheless, these studies reveal that untreated AIH had a very poor prognosis and 5- and 10-year survival rates of 50% and 10% were reported. They furthermore demonstrated that immunosuppressive treatment significantly improved survival.
Up to 30% of adult patients had histological features of cirrhosis at diagnosis. In 17% of patients with periportal hepatitis, cirrhosis developed within five years, but cirrhosis develops in 82% when bridging necrosis or necrosis of multiple lobules is present. The frequency of remission (86%) and treatment failure (14%) are comparable in patients with and without cirrhosis at presentation. Importantly, the presence of cirrhosis does not influence 10-year survival and those patients require a similarly aggressive treatment strategy (Geall 1968, Soloway 1972).
Almost half of the children with AIH already have cirrhosis at the time of diagnosis. Long-term follow-up revealed that few children can completely stop all treatment and about 70% of children receive long-term treatment (Homberg 1987, Gregorio 1997). Most of these patients relapse when treatment is discontinued, or if the dose of the immunosuppressive drug is reduced. About 15% of patients develop chronic liver failure and are transplanted before the age of 18 years.
In elderly patients, a more severe initial histological grade has been reported (Strassburg 2006). The risk of hepatocellular carcinoma varies considerably between the different diseases PBC, PSC and AIH. Particular PSC is regularly complicated by cholangiocarcinoma, gall bladder carcinoma and rarely hepatocellular carcinoma (Zenousi 2014). In contrast, occurrence of HCC in patients with AIH is a rare event and develops only in long-standing cirrhosis.
Who requires treatment?
Autoimmune hepatitis (AIH) is a remarkably treatable chronic liver disease (Manns 2001, Czaja 2010). Untreated, the prognosis of active AIH is dismal, with 5- and 10-year survival rates between 50 and 10% and a well-recognised therapeutic effect exemplified by the last placebo-controlled treatment trials (Soloway 1972, Kirk 1980). For these reasons the indication for treatment is given in any patient who has an established AIH diagnosis, elevations of aminotransferase activities (ALT, AST), an elevation of serum IgG and histological evidence of interface hepatitis or necroinflammatory activity. This has been discussed in the newest version of the AASLD (Manns 2010a) and the EASL (EASL 2015) AIH guidelines. An initial liver biopsy is recommended for confirmation of the diagnosis and for grading and staging. Biopsies are also helpful for observation of aminotransferase activities in serum reflecting inflammatory activity in the liver, which is not always closely correlated.
Who does not require treatment?
Very few patients with an established AIH diagnosis should not be treated. Rare cases, in which the initiation of standard therapy should be weighed against potential side effects, are contraindications with steroids or azathioprine, or for certain other immunosuppressants (see below). In decompensated liver cirrhosis of patients on the waiting list for liver transplantation and in individuals with complete cirrhosis and absent inflammatory activity treatment does not appear beneficial (Manns 2010a, EASL 2015).
Standard treatment strategy
Independent of the clinically- or immunoserologically-defined type of AIH, standard treatment is implemented with predniso(lo)ne alone or in combination with azathioprine. Both strategies are as effective (Manns 2001, Manns 2010a). The basic premise is based upon the findings of studies of almost three decades ago that indicated the effectiveness of steroids in AIH. Since that time, no single multicentre randomised treatment trial in AIH patients has been performed. Advances of alternative treatments are based on small cohorts and on the need to develop strategies for difficult-to-treat patients. The use of prednisone or its metabolite prednisolone, which is used more frequently in Europe, is effective since chronic liver disease does not seem to have an effect on the synthesis of prednisolone from prednisone. The exact differentiation between viral infection and autoimmune hepatitis is important. Treatment of replicative viral hepatitis with corticosteroids must be prevented as well as administration of interferon in AIH, which can lead to dramatic disease exacerbation.
Standard induction treatment and suggested follow-up examinations are summarised in Table 2. Please note the differences in preferred regimen in Europe and the US, which are delineated in the AASLD AIH Guideline (Manns 2010a). Therapy is usually administered over the course of two years. The decision between monotherapy and combination therapy is guided principally by side effects. Long-term steroid therapy leads to cushingoid side effects. Cosmetic side effects decrease patient compliance considerably (Table 3). Serious complications such as steroid diabetes, osteopenia, aseptic bone necrosis, psychiatric symptoms, hypertension and cataract formation also have to be anticipated in long-term treatment. Side effects are found in 44% of patients after 12 months and in 80% of patients after 24 months of treatment. However, predniso(lo)ne monotherapy is possible in pregnant patients. Azathioprine, on the other hand, leads to a decreased dose of prednisone. It bears a theoretical risk of teratogenicity. In addition, abdominal discomfort, nausea, cholestatic hepatitis, rash and leukopenia can be encountered. These side effects are seen in 10% of patients receiving a dose of 50 mg per day. From a general point of view, a postmenopausal woman with osteoporosis, hypertension and elevated blood glucose would be a candidate for combination therapy. In young women, pregnant women or patients with haematological abnormalities, prednisone monotherapy may be the treatment of choice.
Table 2. Treatment regimen and follow-up examinations of autoimmune hepatitis regardless of autoantibody type| Monotherapy | Combination therapy | |||||
| Prednis(ol)-one | 60 mg reduction by 10 mg/week to maintenance of 20 mg/wk reduction by 5 mg to 10 mg find lowest dose in 2.5 mg decrements | 30–60 mg reduction as in monotherapy | ||||
| Azathioprine | n.a. (maintenance with azathioprine: monotherapy: 2 mg/kg body weight) | 1 mg/kg of body weight (Europe) 50 mg (US) | ||||
| Examination | Before therapy | During therapy before remission q 4 weeks | Remission on therapy q 3–6 months | Cessation of therapy q 3 weeks (x 4) | Remission post-therapy q 3–6 months | Evaluation of relapse |
| Physical | + | + | + | + | + | |
| Liver biopsy | + | (+/-) | + | |||
| Blood count | + | + | + | + | + | |
| Aminotrans- ferases | + | + | + | + | + | + |
| Gamma glutamyl- transferase | + | + | + | |||
| Gamma- globulin | + | + | + | + | + | + |
| Bilirubin | + | + | + | + | + | + |
| Coagulation studies | + | + | + | + | + | |
| Autoanti- bodies | + | +/- | + | |||
| Thyroid function tests | + | +/- | + | |||
| Prednis(ol)one | Azathioprine |
| acne moon-shaped face striae rubra dorsal hump obesity weight gain diabetes mellitus cataracts hypertension | nausea vomiting abdominal discomforts hepatotoxicity rash leukocytopenia teratogenicity (?) oncogenicity (?) |
One of the most important variables for treatment success is adherence. The administration of treatment is essential since most cases of relapse are the result of erratic changes of medication and/or dose. Dose reduction is aimed at finding the individually appropriate maintenance dose. Since histology lags 3 to 6 months behind the normalisation of serum parameters, therapy has to be continued beyond the normalisation of aminotransferase levels. Usually, maintenance doses of predniso(lo)ne range between 10 and 2.5 mg. After 12 to 24 months of therapy predniso(lo)ne can be tapered over the course of 4 to 6 weeks to test whether a sustained remission has been achieved. Tapering regimens aiming at withdrawal should be attempted with great caution and only after obtaining a liver biopsy that demonstrates a complete resolution of inflammatory activity. Relapse of AIH and risk of progression to fibrosis is almost universal when immunosuppression is tapered in the presence of residual histological inflammation. Withdrawal should be attempted with caution to prevent recurrence and subsequent fibrosis progression and should be discussed with the patient and closely monitored.
Outcomes of standard therapy can be classified into four categories: remission, relapse, treatment failure and stabilisation.
Remission is a complete normalisation of all inflammatory parameters including histology. The achievement of aminotransferase activities within two-fold of the upper limit of normal is not recommended as treatment goal, rather, normalisation should be aimed at. Remission is ideally the goal of all treatment regimens and ensures the best prognosis. Remission can be achieved in 65 to 75% of patients after 24 months of treatment. Remission can be sustained with azathioprine monotherapy of 2 mg/kg bodyweight (Johnson 1995). This prevents cushingoid side effects. However, side effects such as arthralgia (53%), myalgia (14%), lymphopenia (57%) and myelosuppression (6%) have been observed. Complete remission is not achieved in about 20% of patients and these patients continue to carry a risk of progressive liver injury.
Relapse is characterised by an increase in aminotransferase levels and the reccurrence of clinical symptoms either while on treatment, following tapering of steroid doses to determine the minimally required dose, or, after a complete withdrawal of therapy. Relapse happens in 50% of patients within six months of treatment withdrawal and in 80% after three years. Relapse is associated with progression to cirrhosis in 38% and liver failure in 14%. Relapse requires reinitiation of standard therapy, consideration of dosing as well as diagnosis, and perhaps a long-term maintenance dose with predniso(lo)ne or azathioprine monotherapy.
Treatment failure characterises a progression of clinical, serological and histological parameters during standard therapy. This is seen in about 10% of patients. In these cases the diagnosis of AIH has to be carefully reconsidered to exclude other etiologies of chronic hepatitis. In these patients experimental regimens can be administered or liver transplantation will become necessary.
Stabilisation is the achievement of a partial remission. Since 90% of patients reach remission within three years, the benefit of standard therapy has to be reevaluated in this subgroup of patients. Ultimately, liver transplantation provides a definitive treatment option.
Treatment of elderly patients
The presentation of acute hepatitis, clinical symptoms of jaundice, abdominal pain and malaise have a high likelihood of attracting medical attention and subsequently leading to the diagnosis of AIH (Nikias 1994). More subtle courses of AIH may not lead to clinically relevant signs and may develop unnoticed other than via routine work-up for other problems or via screening programmes. The question of disease onset in terms of initiation of immune-mediated liver disease versus the clinical consequences that become noticeable after an unknown period of disease progression is not easily resolved. Thus, “late onset” AIH may simply just reflect a less severe course of the disease with slower progression to cirrhosis. While LKM positive patients display a tendency towards an earlier presentation, both acute and subtle (earlier and late presentation) variants appear to exist in ANA positive AIH. In practice, the diagnostic dilemma is that AIH is still perceived by many as a disease of younger individuals and that therefore this differential diagnosis is less frequently considered in elderly patients with cryptogenic hepatitis or cirrhosis. Another relevant question resulting from these considerations is the issue of treatment. Standard therapy in AIH consists of steroids alone or a combination with azathioprine. In maintenance therapy azathioprine monotherapy can also be administered but induction with azathioprine alone is not effective. From a general standpoint most internists will use caution when administering steroids to elderly patients, especially in women in whom osteopenia or diabetes may be present.
Recommendations for the treatment of AIH suggest that side effects be weighed against the potential benefit of therapy, and that not all patients with AIH are good candidates for steroid treatment (Manns 2001). Controversy exists surrounding the benefit of therapy in this group of elderly patients. One cohort reported on 12 patients aged over 65 out of a total of 54 AIH patients. Cirrhosis developed after follow-up in 26% irrespective of age although the histological grade of AIH activity was more severe in the elderly group. Although 42% of the patients over 65 did not receive therapy, deaths were only reported in the younger group (Newton 1997). Another cohort of 20 patients aged over 65, reported a longer time to diagnosis (8.5 vs. 3.5 months) with patients presenting mainly with jaundice and acute onset AIH but that they showed a comparable response rate to immunosuppression to that of younger patients (Schramm 2001). The authors also noted that the prevalence of the HLA A1-B8 allotype was less frequent in older patients suggesting a role for immunogenetics.
This point was further elaborated by a report analysing 47 patients with ANA positive AIH aged 60 years and older, as well as 31 patients aged 30 years and younger in whom DR4+/DR3– prevalence was 47% (older) versus 13% (younger) patients (Czaja 2006). Steroid responsiveness was better in the older patients, in line with previous findings in the same cohort (Czaja 1993). Cirrhosis and extrahepatic immune-mediated syndromes including thyroid and rheumatologic disease (47% vs. 26%) were more prevalent in older AIH patients. However, although more treatment failures were observed in the younger patients (24% vs 5%), the rates of remission, sustained remission and relapse were similar. Interestingly, an assessment of age-stratified prevalence showed an increase after the age of 40 from 15% to over 20%.
From all this data, AIH in elderly patients appears to be characterised by a distinct clinical feature, a distinct immunogenetic profile, favourable response rates and higher rates of cirrhosis present at diagnosis, all of which contribute to the heterogeneity of AIH. A UK cohort of 164 AIH patients included 43 individuals aged 60 years (Al-Chalabi 2006). The different age groups showed no significant differences regarding serum biochemistry, autoantibody titres, time to establishment of diagnosis, and mode of presentation. The authors provided a substratification of patients below and above 40 years of age and reported that older patients had a higher median histological stage and a comparable median grade but that younger patients had more median relapse episodes and a higher median stage at follow-up biopsy. The most distinguishing clinical sign was a higher prevalence of ascites in the older group. However, rates of complete, partial and failed response were similar, and the median number of relapses was higher in younger patients, which nevertheless did not lead to differences in liver-related deaths in either group (12% vs. 15%). In comparison to the study of ANA positive AIH patients from the US (Czaja 2006), the differing findings regarding HLA association are noteworthy. In the UK study there was no differential distribution of HLA DR3 and DR4 and this questions the suggested hypothesis of a primary influence of immunogenetics on the observed clinical distinctions. The reasons for the clinical differences of AIH in older and younger patients are unclear. They may include differences in hepatic blood flow and alterations involving the regulation of cellular immunity during ageing (Talor 1991, Prelog 2006). In summary, these data suggest that AIH in elderly patients should be considered and treated (Strassburg 2006).
Alternative treatments
When standard treatment fails or drug intolerance occurs, alternative therapies such as cyclosporine, tacrolimus, cyclophosphamide, mycophenolate mofetil, rapamycin, UDCA, and budesonide can be considered (Table 4). The efficacy of most of these options has not yet been definitively decided and is only reported in small case studies.
Budesonide
Budesonide is a synthetic steroid with high first-pass metabolism in the liver, in principle with limited systemic side effects compared to conventional steroids. In comparison to prednisone the absolute bioavailability of budesonide is less than 6-fold lower (Thalen 1979) but it has an almost 90% first-pass metabolism in the liver, a higher affinity to the glucocorticoid receptor, acts as an anti-inflammatory and immunosuppressive drug and leads to inactive metabolites (6-OH-budesonide, 16-OH-prednisolone). In a pilot study treating 13 AIH patients with budesonide over a period of 9 months the drug was well-tolerated and aminotransferase levels were normalised (Danielson 1994). However, in a second study budesonide therapy was associated with a low frequency of remission and high occurrence of side effects (Czaja 2000) in 10 patients who had previously been treated with azathioprine and steroids and had not reached a satisfactory remission. This study concluded that budesonide was not a good treatment option in those patients. A third study reported that remission was induced with budesonide combination therapy in 12 previously untreated patients (Wiegand 2005). The authors performed kinetic analyses and reported that the area under the curve (AUC) of budesonide was increased in those with high inflammatory activity and cirrhosis. This finding plausibly demonstrates that in patients with portosystemic shunts in portal hypertension the effect of high hepatic first-pass metabolism that would limit typical steroid side effects is reduced.
Table 4. Alternative drugs in autoimmune hepatitis| Compound | Advantage | Disadvantage |
| Budesonide | High first pass effect Immunosuppressive action Inactive metabolites | Cirrhosis (portosystemic shunts) and side effects |
| Cyclosporine | Satisfactory experience Potent immunosuppressant Transplant immunosuppressant | Renal toxicity |
| Tacrolimus | Potent immunosuppressant Transplant immunosuppressant | Renal toxicity |
| Mycophenolic acid | Favourable toxicity profile Transplant immunosuppressant | Disappointing effectiveness |
| Cyclophosphamide | Effective | Continuous therapy Hematological side effects |
The main advantage of budesonide for the future treatment of autoimmune hepatitis would therefore be to replace prednisone in long-term maintenance therapy and induction therapy to reduce steroid side effects. To this end the first multicentre placebo-controlled randomised AIH treatment trial in 3 decades was performed with a total of 207 non-cirrhotic patients from 30 centres in nine European countries and Israel (Manns 2010b). In this trial 40 mg prednisone (reduction regimen) and azathioprine was compared to 3 mg budesonide (TID initially, reduced to BID) in combination with azathioprine. The data shows that budesonide in combination with azathioprine is efficient in inducing stable remission, is superior in comparison to a standard prednisone tapering regimen beginning with 40 mg per day and leads to a substantially superior profile of steroid-specific side effects. From these data, budesonide has emerged as an alternative first line treatment strategy for non-cirrhotic patients with AIH (Manns 2010b, EASL 2015). Budesonide is licensed for the use in AIH in many countries. Effective treatment of children with budesonide has been reported (Woynarowski 2013).
Deflazacort
This alternative corticosteroid has also been studied for immunosuppression in AIH because of its feature of fewer side effects than conventional glucocorticoids. In a pilot study 15 patients with AIH type 1 were treated with deflazacort, who had been previously treated with prednisone with or without azathioprine until they reached a biochemical remission. Remission was sustained for two years of follow-up. However, the long-term role of second-generation corticosteroids to sustain remission in AIH patients with reduced treatment-related side effects requires further controlled studies (Rebollo Bernardez 1999).
Cyclosporine A
Cyclosporine A (CyA) is a lipophylic cyclic peptide of 11 residues produced by Tolypocladium inflatum that acts on calcium-dependent signaling and inhibits T cell function via the interleukin 2 gene (Strassburg 2008). Out of the alternative AIH drugs considerable experience has been reported with CyA. CyA was successfully used for AIH treatment and was well tolerated (Alvarez 1999b, Debray 1999). The principal difficulty in advocating widespread use of CyA as first line therapy relates to its toxicity profile, particularly with long-term use (increased risk of hypertension, renal insufficiency, hyperlipidaemia, hirsutism, infection, and malignancy) (Alvarez 1999b, Debray 1999, Fernandez 1999, Heneghan 2002).
Tacrolimus
Tacrolimus is a macrolide lactone compound with immunosuppressive qualities exceeding those of CyA. The mechanism of action is similar to that of CyA but it binds to a different immunophilin (Strassburg 2008). The application of tacrolimus in 21 patients treated for one year led to an improvement of aminotransferase and bilirubin levels with a minor increase in serum BUN and creatinine levels (Van Thiel 1995). In a second study with 11 steroid-refractory patients, improvement of inflammation was also observed (Aqel 2004). A recent study demonstrated the effectiveness of tacrolimus in difficult to treat patients (Than 2016). However, although tacrolimus represents a promising immunosuppressive candidate drug, larger randomised trials are required to assess its role in the therapy of AIH.
Mycophenolic acid
Mycophenolate is a noncompetitive inhibitor of inosine monophosphate dehydrogenase, which blocks the rate-limiting enzymatic step in de novo purine synthesis and is widely used in solid organ transplantation. Mycophenolate has a selective action on lymphocyte activation, with marked reduction of both T and B lymphocyte proliferation. In a pilot study, seven patients with AIH type 1 who either did not tolerate azathioprine or did not respond to standard therapy with a complete normalisation of aminotransferase levels, were treated with mycophenolate in addition to steroids. Normalisation of aminotransferase levels was achieved in five out of seven patients within three months. These preliminary data suggested that mycophenolate may represent a promising treatment strategy for AIH (Richardson 2000). However, in a retrospective study, there was no statistically significant benefit for mycophenolate treatment in 37 patients with AIH and azathioprine failure or intolerance who were treated with mycophenolate (Hennes 2008a). Less than 50% reached remission and in the azathioprine non-responders failure was 75%. Mycophenolate has been demonstrated to be most effective as a second line therapy in patients found to be intolerant to azathioprine. There is some evidence that mycophenolate can be used as first line therapy (Zachou 2016). There is limited data available on the use of mTOR inhibitors such as everolimus in AIH (Ytting 2015).
Cyclophosphamide
The induction of remission with 1–1.5 mg per kg per day of cyclophosphamide in combination with steroids has been reported. However, the dependency of continued application of cyclophosphamide with its potentially severe haematological side effects renders it a highly experimental treatment option (Kanzler 1996).
Anti-TNF α antibodies
There is some emerging evidence that anti-TNF antibodies are capable of inducing remission in AIH patients in whom standard or alternative therapeutic options have been exhausted (Efe 2010, Umekita 2011, Weiler-Norman 2013). However, the development of AIH has also been observed under treatment with anti-TNF antibodies (Ramos-Casals 2008). Future studies will have to define the role of this therapeutic option in difficult-to-treat cases of AIH.
Ursodeoxycholic acid
Ursodeoxycholic acid is a hydrophilic bile acid with putative immunomodulatory capabilities. It is presumed to alter HLA class I antigen expression on cellular surfaces and to suppress immunoglobulin production. Uncontrolled trials have shown a reduction in histological abnormalities, clinical and biochemical improvement but not a reduction of fibrosis in four patients with AIH type 1 (Calmus 1990, Nakamura 1998, Czaja 1999). However, its role in AIH therapy or in combination with immunosuppressive therapy is still unclear.
Other alternative treatment strategies include methotrexate, anti-TNF α antibodies, and rituximab, but there is currently insufficient data on any of these.
Overlap syndromes and treatment
Overlap syndrome describes a disease condition that is not completely defined (Strassburg 2006). A valid definition is difficult (Boberg 2011). It is characterised by the coexistence of clinical, biochemical or serological features of autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and depending on the definition, also viral hepatitis C (HCV) (Ben-Ari 1993, Colombato 1994, Duclos-Vallee 1995, Chazouilleres 1998, Angulo 2001, Rust 2008). In adult patients an overlap of PBC and AIH is most frequently encountered although it is unclear whether this is true co-existence of both diseases or an immunoserological overlap characterised by the presence of antinuclear (ANA) as well as antimitochondrial (AMA) antibodies (Poupon 2006, Gossard 2007, Silveira 2007, Al-Chalabi 2008). In many AMA negative patients with a cholestatic liver enzyme profile ANA are present. This has been termed autoimmune cholangiopathy or AMA negative PBC (Michieletti 1994).
Apart from coexisting, autoimmune liver diseases can also develop into each other, i.e., the sequential manifestation of PBC and autoimmune hepatitis. The true coexistence of AIH and PSC has only been conclusively shown in paediatric patients (Gregorio 2001). It can be hypothesised whether a general predisposition toward liver autoimmunity exists which has a cholestatic, a hepatitic and a bile duct facet, which may be variable depending upon unknown host factors. The diagnosis of an overlap syndrome relies on the biochemical profile (either cholestatic with elevated alkaline phosphatase, gamma glutamyltransferase and bilirubin, or hepatitic with elevated aspartate aminotransferase and alanine aminotransferase levels in addition to elevated gamma globulins), the histology showing portal inflammation with or without the involvement of bile ducts, and the autoantibody profile showing AMA or autoantibodies associated primarily with AIH such as liver-kidney microsomal antibodies (LKM), soluble liver antigen antibodies (SLA/LP) or ANA. In cholestatic cases cholangiography detects sclerosing cholangitis. In an overlap syndrome the classical appearance of the individual disease component is mixed with features of another autoimmune liver disease. Immunoglobulins are usually elevated in all autoimmune liver diseases.
Regarding a therapeutic strategy, the leading disease component is treated. In an overlap syndrome presenting as hepatitis, immunosuppression with prednisone (or combination therapy with azathioprine) is initiated. In cholestatic disease ursodeoxycholic acid is administered. Both treatments can be combined when biochemistry and histology suggest a relevant additional disease component (Chazouilleres 1998). Validated therapeutic guidelines for overlap syndromes are not available. It is important to realise that treatment failure in AIH may be related to an incorrect diagnosis or an overlap syndrome of autoimmune liver diseases (Potthoff 2007). Several studies show that treatment of the AIH component of overlap syndromes is important to avoid progression to cirrhosis (Chazouilleres 2006, Gossard 2007, Silveira 2007, Al-Chalabi 2008).
Liver transplantation
In approximately 10% of AIH patients liver transplantation remains the only life-saving option (Strassburg 2004). The indication for liver transplantation in AIH is similar to that in other chronic liver diseases and includes clinical deterioration, development of cirrhosis, bleeding oesophageal varices and coagulation abnormalities despite adequate immunosuppressive therapy (Neuberger 1984, Sanchez-Urdazpal 1991, Ahmed 1997, Prados 1998, Tillmann 1999, Vogel 2004). There is no single indicator or predictor for the necessity of liver transplantation. Candidates for liver transplant are usually patients who do not reach remission within four years of continuous therapy. Indicators of a high mortality associated with liver failure are histological evidence of multilobular necrosis and progressive hyperbilirubinaemia. In Europe, 4% of liver transplants are for AIH (Strassburg 2009). The long-term results of liver transplantation for AIH are excellent. The five-year survival is up to 92% (Sanchez-Urdazpal 1991, Prados 1998, Ratziu 1999) and well within the range of other indications for liver transplantation. The European liver transplant database indicates 76% survival in five years and 66% survival after 10 years (1647 liver transplantations between 1988 and 2007). When these numbers are considered it is necessary to realise that patients undergoing liver transplantation usually fail standard therapy and may therefore have a reduced life expectancy after liver transplant compared to those who achieve stable complete remission on drug therapy.
Recurrence and de novo AIH after liver transplantation
The potential of AIH to recur after liver transplantation is beyond serious debate (Schreuder 2009). The first case of recurrent AIH after liver transplant was reported in 1984 (Neuberger 1984) and was based upon serum biochemistry, biopsy findings and steroid reduction. Studies published over the years indicate that the rate of recurrence of AIH ranges between 10–35%, and that the risk of AIH recurrence is perhaps as high as 68% after five years of follow-up (Wright 1992, Devlin 1995, Götz 1999, Milkiewicz 1999, Manns 2000, Vogel 2004). It is important to consider the criteria upon which the diagnosis of recurrent AIH is based. When transaminitis is chosen as a practical selection parameter many patients with mild histological evidence of recurrent AIH may be missed. It is therefore suggested that all patients with suspected recurrence of autoimmune hepatitis receive a liver biopsy, biochemical analyses of aminotransferases as well as a determination of immunoglobulins and autoantibody titres (Vogel 2004). Significant risk factors for the recurrence of AIH have not yet been identified although it appears that the presence of fulminant hepatic failure before transplantation protects against the development of recurrent disease. Risk factors under discussion include steroid withdrawal, tacrolimus versus cyclosporine, HLA mismatch, HLA type, and LKM-1 autoantibodies. An attractive risk factor for the development of recurrent AIH is the presence of specific HLA antigens that may predispose toward a more severe immunoreactivity. In two studies recurrence of AIH appeared to occur more frequently in HLA DR3 positive patients receiving HLA DR3 negative grafts. However, this association was not confirmed in all studies. There have not been conclusive data to support the hypothesis that a specific immunosuppressive regimen represents a risk factor for the development of recurrent AIH (Gautam 2006). However, data indicate that patients transplanted for AIH require continued steroids in 64% versus 17% of patients receiving liver transplants for other conditions (Milkiewicz 1999).
Based on these results and other studies it would appear that maintenance of steroid medication in AIH patients is indicated to prevent not only cellular rejection but also graft-threatening recurrence of AIH (Vogel 2004). Steroid withdrawal should therefore be performed only with great caution. The recurrence of AIH is an important factor for the probability of graft loss. Apart from HCV and primary sclerosing cholangitis a recent report found AIH recurrence to represent the third most common reason for graft loss (Rowe 2008). Transplanted patients therefore require a close follow-up and possibly an immunosuppressive regimen including steroids, although this is controversial and not backed by prospective studies (Campsen 2008).
In addition to AIH recurrence the development of de novo autoimmune hepatitis after liver transplantation has been reported (Kerkar 1998, Jones 1999a, Salcedo 2002). The pathophysiology of this is also elusive. From a treatment point of view de novo autoimmune hepatitis, which appears to occur mostly in patients transplanted with PBC but may just be the serendipitous occurrence of AIH, is responsive to steroid treatment (Salcedo 2002).
Primary biliary cholangitis
Introduction
The former designation“primary biliary cirrhosis” is no longer used because it labels patients as having cirrhosis where this is often not the case. However, the acronym PBC remains unchanged (Beuers 2015). PBC is a chronic inflammatory, cholestatic disease of the liver with an unknown cause. The clinical observation of a broad array of immune-mediated symptoms and phenomena suggests the disease to be of autoimmune aetiology, in the course of which progressive and irreversible destruction of small interlobular and septal bile ducts progressively and irreversibly ensues (Table 5). As in other autoimmune diseases PBC affects women in over 80% of cases and is associated with varying extrahepatic autoimmune syndromes in up to 84%. These extrahepatic manifestations of immune-mediated disease include the dry gland syndrome (sicca syndrome with xerophthalmia and xerostomia) but also collagen diseases, autoimmune thyroid disease, glomerulonephritis and ulcerative colitis (Table 6).
Table 5. Clinical profile of primary biliary cholangitis (PBC)| Sex | 90% female |
| Age | 40–59 yrs pruritus jaundice skin pigmentation |
| Elevation | alkaline phosphatase (AP), aspartate aminotransferase (AST), bilirubin, IgM antimitochondrial antibodies (AMA) associated immune-mediated syndromes |
| Liver biopsy | cellular bile duct infiltration granulomas possible copper deposits |
The striking female predominance (Donaldson 1996, Mackay 1997, Uibo 1999) and familiar clustering of PBC (Kato 1981, Jones 1999b, Tsuji 1999) suggest that inheritable genetic factors play a role in this disease. This has focused attention on the immunogentics of PBC in order to further define host risk factors (Manns 1994). Studies have suggested an instability of lymphocytic DNA in PBC patients (Notghi 1990). Immunogentic analyses, however, have only come up with relatively weak associations with specific human leukocyte antigen haplotypes. An additional hypothesis is an alteration of bile acid composition and bile fluid composition, which would indicate a role for transporter proteins in the development of PBC. Bicarbonate rich bile is believed to be protective for biliary epithelium.
Table 6. Extrahepatic immune-mediated syndromes in PBC and overlap with rheumatic diseases| Dry gland “sicca” syndrome |
| Sjögren’s syndrome |
| Rheumatoid arthritis |
| Autoimmune thyroid disease |
| Renal tubular acidosis |
| Mixed connective tissue disease (MCTD) |
| Polymyositis |
| Polymyalgia rheumatic |
| Pulmonary fibrosis |
| CREST syndrome |
| Systemic lupus erythematosus (SLE) |
| Pernicious anaemia |
| Ulcerative colitis |
| Exogenous pancreatic insufficiency |
| Myasthenia gravis |
Definition and prevalence of PBC
PBC is an inflammatory, primarily T cell-mediated chronic destruction of intrahepatic microscopic bile ducts of unknown aetiology (Strassburg 2000). It affects women in 80% of cases who exhibit elevated immunoglobulin M, antimitochondrial antibodies directed against the E2 subunit of pyruvate dehydrogenase (PDH-E2), a cholestatic liver enzyme profile with elevated alkaline phosphatase, gamma glutamyltransferase as well as serum bilirubin levels, and a variable course of disease leading to cirrhosis over the course of years or decades. A prominent feature is the presence of extrahepatic immune-mediated disease associations. In later stages, pronounced fatigue, pruritus, marked hyperbilirubinaemia and the consequences of portal hypertension such as ascites, bleeding oesophageal varices, and encephalopathy develop (Strassburg 2004).
The prevalence is estimated at 65 per 100,000 in women and 12 per 100,000 in men with an incidence of 5 per 100,000 in women and 1 per 100,000 in men. The prevalence and incidence appear to vary regionally and appears to be increasing (Boonstra 2012). An increase of PBC incidence in recent years may be the result of more specific testing of antimitochondrial antibody reactivity (Strassburg 2004).
Diagnostic principles of PBC
Suspicion of PBC arises when cholestasis and cirrhosis are present in middle-aged women (Figure 2). Ultrasound is employed to rule out mechanical cholestasis. The presence of antimitochondrial antibodies (AMA) against PDH-E2 is diagnostic of PBC. AMA against E2 subunits of members of the inner mitochondrial membrane-expressed oxoacid dehydrogenase complex (PDH, branched chain ketoacid dehydrogenase [BCKD], and ketoglutarate dehydrogenase [OADC]) are present in 95% of PBC patients. AMA negative PBC can exhibit antinuclear autoantibodies with specificity against nuclear dot antigen (SP100), a 210 kDa nuclear membrane protein (gp210), or nucleoporin p62. In AMA negative PBC a biopsy is indicated to contribute to the establishment of the diagnosis; in the presence of AMA against PDH-E2, histology is used primarily for the staging of cirrhosis and is not necessary (Strassburg 2004). The diagnosis is established when 2 of the main criteria (cholestatic biochemistry, AMA or PBC-specific autoantibody, typical histology) are met.
Diagnostic role of AMA in PBC
The main aim of AMA determinations is the detection of PBC-specific AMA and the exclusion of AMA of low diagnostic relevance for the disease. As a screening test the determination of AMA using indirect immunofluorescence testing on rat kidney cryostat sections or immobilised Hep-2 cells (Strassburg 1999). The indirect immunofluorescence on rat kidney sections leads to the staining of the distal and proximal tubuli (note: proximal staining only is indicative of liver/kidney microsomal antibodies, LKM). When positive AMA immunofluorescence is detected, further analysis should include subclassification using molecularly defined antigen preparations. The detection of PDH-E2, BCKD-E2 can be achieved by ELISA using recombinant antigen or reference sera. If both are negative, testing should include OGD-E2. The final step is performed using western blot Analyses to confirm the findings. By western blot the indicative 74 kDa (PDH-E2), 52 kDa (BCKD-E2) and 48 kDa (OGD-E2) bands can be visualised. This multi-step regimen secures a rational and reliable diagnosis of PBC-specific AMA excluding those found in drug-induced and infectious diseases.
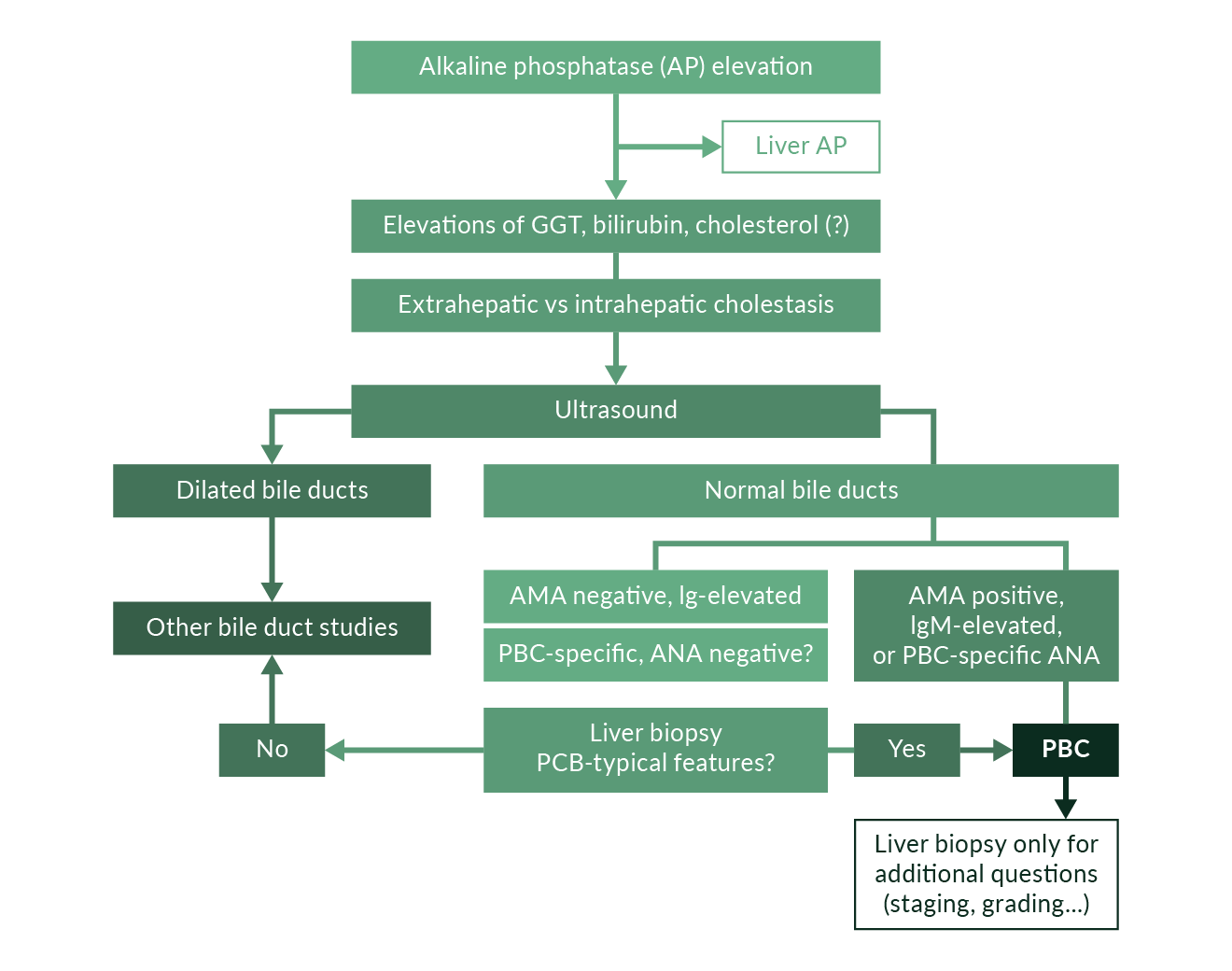 Figure 2. Diagnostic algorithm of PBC including clinical presentation, ultrasound and serology
Figure 2. Diagnostic algorithm of PBC including clinical presentation, ultrasound and serology
In the majority of cases the determination of anti-PDH-E2 is sufficient to secure the diagnosis. Studies will have to evaluate whether the future application of a single PDH-E2 ELISA as highly specific screening test in suspected PBC represents an efficient and economic diagnostic approach.
Therapeutic principles in PBC
There is currently no cure for PBC (Strassburg 2004). Ursodeoxycholic acid (UDCA) (15 mg/kg body weight per day) has been shown to improve serum biochemistry, histology and survival but has no effect on fatigue and osteoporosis. It has immunomodulatory properties, alters cell signal transduction and modifies hydrophilicity of the bile. UDCA should not be given in severe cholestasis and during the first trimester of pregnancy. Immunosuppression in PBC has shown disappointing results. Symptomatic therapy of the complications of PBC includes management of pruritus (cholestyramine, induction with rifampicin, opioid antagonists, serotonin antagonists), ascites (diuretics, beta blockers to control portal hypertension), osteoporosis (vitamin D and calcium supplementation, bisphosphonates in some), as well as endoscopic intervention for bleeding oesophageal varices. Fat-soluble vitamin replacement is suggested. When liver cirrhosis-induced liver failure is progressive, liver transplantation remains a definitive therapeutic option. Ten-year survival rates are 75–80% and recurrence of PBC after transplant occurs in 10 to 40% of patients. Recurrence can be expected in 25 to 30% (Rowe 2008, Strassburg 2009). The number of PBC patients on the waiting lists has declined during the past decade. The risk for death on the waiting list in PBC patients with jaundice is significantly higher than in those patients with HCV infection or alcoholic liver cirrhosis.
Immunosuppression in PBC
Corticosteroids: Treatment with prednisolone can improve serum aminotransferase activities, alkaline phosphatase and elevated immunoglobulins. It does not lead to significant improvement of bilirubin, pruritus, or histology. In a placebo-controlled study with 36 asymptomatic patients for over one year osteopenia and cushingoid side effects were noted (Mitchison 1992).
Azathioprine: The classical immunosuppressant azathioprine, which has a pronounced effect in AIH, did not show significant effects in two different studies and is not used in PBC (Christensen 1985).
Cyclosporine A: In a large study of 346 patients with a median observation time of 2.5 years, this classical transplant immunosuppressant did not show significant effects on histological progression (Lombard 1993). Histology did improve in a small study with 20 patients who were treated for two years, but these results should be viewed with caution (Wiesner 1990). Because of the possibility of severe side effects, cyclosporin A is not a recommended therapeutic option.
D-penicillamine: Because PBC is characterised by copper accumulation in the bile ducts the chelator d-penicillamine was studied. D-penicillamine also has immunosuppressive and antifibrotic properties. It was tested on a total of 748 patients in six studies, without leading to a positive therapeutic effect while 30% of patients had severe side effects (Bodenheimer 1985). D-penicillamine in PBC is not recommended.
Colchicine: Because of its antifibrotic and anti-inflammatory properties colchicine was studied in the 1980s. Despite improvement of albumin, bilirubin, aminotransferases and alkaline phosphatase, an improvement of clinical symptoms and histology was not observed (Kaplan 1986, Warnes 1987, Bodenheimer 1988). Severe side effects were not reported but an effect on long-term prognosis was not seen.
Methotrexate: Despite its known hepatotoxicity, methotrexate was used as an immunosuppressant in PBC. In a placebo-controlled study with 60 patients, low-dose methotrexate (7.5 mg/week) led to an improvement of biochemical parameters except for bilirubin but no effects were reported regarding necessity of liver transplantation or survival (Hendrickse 1999). Hepatotoxicity was not observed. Interstitial pneumonitis, which affects 3–5% of rheumatoid arthritis patients, was observed in 14% of PBC patients. Methotrexate cannot be recommended outside of scientific evaluations or studies.
In principle, other immunosuppressants (Table 7) such as mycophenolic acid (mycophenolate mofetil), tacrolimus (FK506) or even monoclonal antibodies against the interleukin 2 receptor may represent interesting candidate strategies. However, data is currently lacking.
Table 7. Effects of immunosuppressants in PBC| Biochemical improvement | Histological improvement | Survival | Side effects/toxicity | |
| Corticosteroids | ++ | ++ | – | ++ |
| Azathioprine | – | – | + | + |
| Cyclosporin A | ++ | – | ++ | ++ |
| D-penicillamine | – | – | – | ++ |
| Colchicine | ++ | - | + | – |
| Methotrexate | ++ | + | – | + |
Ursodeoxycholic acid in PBC (UDCA)
In 1981, a positive effect of UDCA was observed on elevated liver parameters, the exact mechanism of which was unclear (Leuschner 1996). On one hand UDCA leads to a modification of the bile acid pool to a more hydrophilic environment with lower detergent-like properties, and it leads to increased bile flow. On the other hand an immunomodulatory activity is suggested regarding HLA antigens expressed on biliary epithelial cells and altered signal transduction (Paumgartner 2002). The optimal dose in PBC patients appears to be 13–15 mg/kg. In a meta-analysis of three studies that looked at 548 patients with this dose, biochemical improvement and a slower histological progression to fibrosis was observed (Poupon 1997). These effects were only evident when follow-up extended to four years. These data rely heavily on the positive effects of a single study and it is not surprising that a subsequent meta-analysis of eight studies with 1114 patients failed to find positive associations with UDCA therapy (Goulis 1999).
There are a number of problems with this analysis. Doses varied and some protocols included patients with insufficient dosing, and follow up was less than two years in some studies. In a recently published analysis of 367 patients from four clinical cohorts, initiation of UDCA therapy in early stages of PBC (stage I-II) and a treatment duration of two years led to a retardation of histological progression, which argues for an early initiation of UDCA therapy after diagnosis, even in the absence of fibrosis or cirrhosis. UDCA was also shown to improve biochemistry, delay portal hypertension and varices, and currently has no therapeutic alternative (Poupon 2003). No convincing effect was demonstrable on osteopenia and extrahepatic manisfestations of PBC. A number of parameters have been studied to assess the prognosis of PBC measured by the observed biochemical response to UDCA therapy. Several criteria have been reported including the Corpechot, Parès, and Rotterdam criteria, which in summary describe the reduction of AST, AP, and bilirubin after one year of UDCA treatment. Currently the prognostic stratification is based upon the assessment of treatment response to UDCA after 12 months. A reduction of AST, AP and bilirubin indicates a favourable outcome of therapy and should be monitored during therapy (Corpechot 2011, Kuiper 2009). Additional predictive scores of UDCA-treated PBC patients are currently being developed and evaluated (Bowlus 2016).
A novel therapeutic strategy involves the use of obeticholic acid (OCA), which is an inducer of the farnesoid-X-receptor (FXR). The first studies have been favourable regarding biochemical response to therapy. Unfortunately OCA is associated with severe pruritus and drug discontinuations in a significant number of patients at doses above 10 mg (Hirschfeld 2014). OCA is also implicated to reduce portal hypertension (Verbeke 2014) and fibrosis (Verbeke 2016), and is a candidate for combined therapy with budesonide acting via the pregnane-X-receptro (PXR) in addition to FXR (Silveira 2014). A phase 3 trial assessing 5–10 mg OCA in combination with UDCA has recently been published demonstrating a decrease of baseline biochemical parameters believed to correlate with prognosis (Nevens 2016). OCA has been licensed for use as second line therapy in the US and Europe. The effects on fibrosis progression remain to be shown in long term observations.
Therapy in non-responders and combination strategies
Non-response is usually defined as a failure to lower cholestatic enzyme activities or to reach normalisation of these parameters (Kuiper 2009). In patients in whom alkaline phosphatase and gamma glutamyltransferase activities are not lowered by UDCA therapy, increased morbidity and progression is likely. Alternative therapeutic strategies can be considered.
Steroids and UDCA: The combination of immunosuppressants and UDCA was looked at in smaller studies and included the use of prednisolone (Leuschner 1996), azathioprine (Wolfhagen 1998) and budesonide (Leuschner 1999, Angulo 2000) (Table 7). In a randomised, controlled study with 30 patients who received 10 mg prednisolone/day an improvement of inflammatory activity was reported (Leuschner 1996). A study with 9 mg budesonide/day in 39 patients showed not only biochemical but also histological improvement (Leuschner 1999). In an open study with 22 patients a deterioration of osteopenia was noted (Angulo 2000). The combination of budesonide and UDCA may have additional beneficial effects related to the activation of the anion exchanger AE2, which may serve to alter biliary composition and produce a more protective bicarbonate rich bile.
Sulindac and UDCA: In an open study with 23 patients and incomplete response to UDCA over 12 months treated with UDCA or UDCA and sulindac a trend towards histological improvement and biochemical improvement were reported in the combination group (Leuschner 2002).
Colchicine and UDCA: Three studies investigated the combination of colchicine and UDCA for 24 months in a total of 118 patients (Raedsch 1992, Ikeda 1996, Poupon 1996). Although mild biochemical improvement was noted, the effect of longer treatment remains unclear. Because of the biliary elimination of colchicine combinations with bile acids, there may be potentially toxic effects.
Methotrexate and UDCA: Several pilot studies and three randomised studies have looked at methotrexate in combination with UDCA. In one randomised placebo-controlled protocol with 60 patients a high rate of side effects without therapeutic benefit was reported (Van Steenbergen 1996, Bach 2003).
Fibrates: An interesting therapeutic approach is the use of fibrates (bezafibrate or fenofibrate) to improve the response to UDCA in non- or partial responders. Fibrates act by induction peroxisome proliferator-activated receptor α (PPARα)-UDP-glucuronosyltransferases (UGTs) signaling axis which is an important determinant of bile acid homeostasis. Beza- or fenofibrate have been studied in 25 studies (Floreani 2016). Results of a large trial with bezafibrate are pending.
Other strategies in future focus on the use of taurine conjugated UDCA (T-UDCA), norUDCA and synthetic PPAR δ agonists.
Primary sclerosing cholangitis
Diagnosis of primary sclerosing cholangitis (PSC)
PSC is classically characterised by the progressive destruction of large intra- as well as extrahepatic bile ducts and – contrasting with AIH and PBC – preferentially affects male patients with a maximum age of around 25 to 45 (Strassburg 1996). About 50 to 75% of the time, PSC is associated with ulcerative colitis. The aetiology of PSC remains elusive but genome-wide association studies have identified susceptibility loci, which share features between PSC and inflammatory bowel disease (Janse 2011, Liu 2013). PSC is clinically characterised by upper quadrant pain, pruritus, anorexia and fever, but up to 50% of patients lack symptoms (Weismüller 2008). The diagnosis is established by a typical biochemical profile of cholestasis with elevations of bilirubin, alkaline phosphatase and gamma glutamyl transferase, the characteristic findings upon cholangiography and a typical biopsy showing ring fibrosis around the bile ducts, which is not present in all patients. Serology regularly identifies atypical anti-neutrophil cytoplasmic autoantibodies (xANCA) in up to 80% of patients (Terjung 2000), although these are not disease-specific and can also occur in patients with ulcerative colitis without PSC. These autoantibodies also occur in bile of PSC patients and correlate with disease activity (Lenzen 2013). There is a significant association of PSC with cholangiocarcinoma (10–20%) and colorectal cancer (9% in 10 years). In a subgroup of patients, small bile duct PSC may be present (Broome 2002), which lacks typical strictures and pruning of the biliary tree upon cholangiography. In these cases the diagnosis can be established in the presence of the typical association with ulcerative colitis in male patients by performing a liver biopsy (Figure 3).
 Figure 3. Diagnostic algorithm of PSC including clinical presentation
Figure 3. Diagnostic algorithm of PSC including clinical presentation
 Figure 4a. Examples of different entities of sclerosing cholangitis. A) PSC showing multiple strictures with narrowing (black arrows) and prestenotic dilatation (white arrows) and an endoscopic aspect of purulent biliary infection at the biliary papilla
Figure 4a. Examples of different entities of sclerosing cholangitis. A) PSC showing multiple strictures with narrowing (black arrows) and prestenotic dilatation (white arrows) and an endoscopic aspect of purulent biliary infection at the biliary papilla
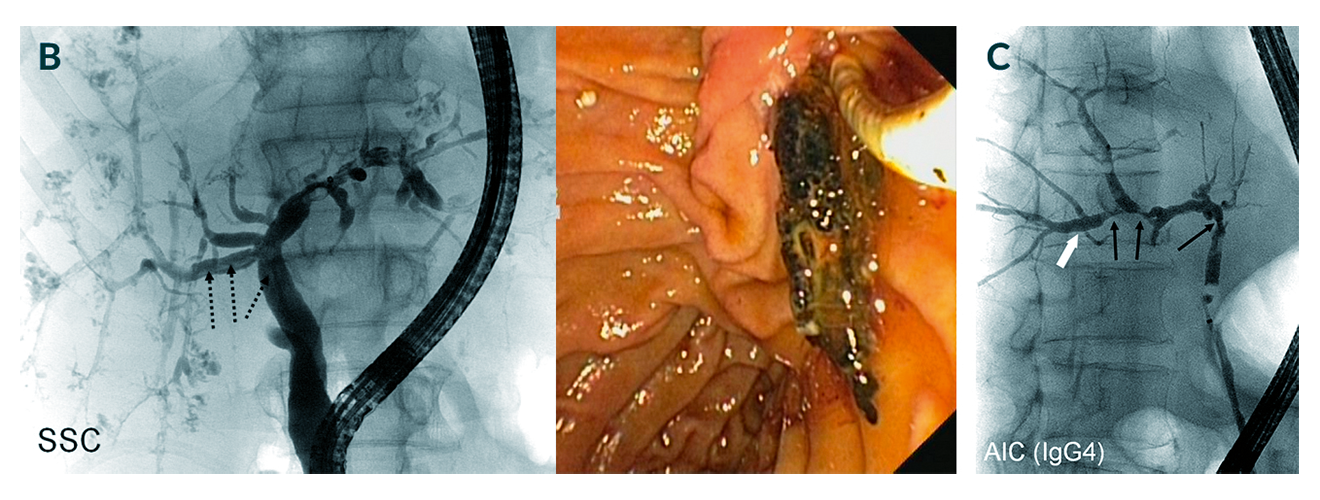 Figures 4b, 4c. Examples of different entities of sclerosing cholangitis. B) Secondary sclerosing cholangitis (SSC) with a similar intrahepatic picture but also biliary casts (dotted arrows) that can be extracted endoscopically (right panel). C) Cholangiogram of autoimmune (AIC) IgG4-associated cholangitis mimicking PSC. Black arrows show narrowing, white arrows show dilatations
Figures 4b, 4c. Examples of different entities of sclerosing cholangitis. B) Secondary sclerosing cholangitis (SSC) with a similar intrahepatic picture but also biliary casts (dotted arrows) that can be extracted endoscopically (right panel). C) Cholangiogram of autoimmune (AIC) IgG4-associated cholangitis mimicking PSC. Black arrows show narrowing, white arrows show dilatations
Differential diagnosis: sclerosing cholangitis
The finding of macroductal sclerosing cholangitis can be brought about by a number of conditions, which include ischaemia, liver transplantation complications, and drugs. The dilemma is that PSC is primarily a visual diagnosis shared by many other entities leading to features of sclerosing cholangitis (Figure 5). Of note are two additional differential diagnoses that require attention (Figure 4): secondary sclerosing cholangitis (Gelbmann 2007, Esposito 2008, von Figura 2009, Al-Benna 2011) and IgG4-associated cholangitis (Webster 2009, Clendenon 2011, Takuma 2011, Zhang 2011).
Secondary sclerosing cholangitis is an entity with severe infection of the biliary tree that develops in some patients following systemic infections and sepsis who are treated with aggressive intensive care unit management. IgG4-associated cholangitis is an immune-mediated systemic disease, which mainly affects the pancreas and bile ducts but also the lymph nodes, the kidneys, the thyroid and many other organs (Kamisawa 2014). It is characterised by often high plasma levels of IgG4 and IgG4 expression in plasma cells obtained upon brush or forceps biopsy. The latter can be treated with immunosuppression and should be diagnosed because of an available medical therapy (Kamisawa 2014).
 Figure 5. Diseases of the liver and those affecting the liver, which can lead to features of sclerosing cholangitis. The differential diagnostic considerations in visually apparent sclerosing cholangitis cover a diverse array of conditions apart from PSC.
Figure 5. Diseases of the liver and those affecting the liver, which can lead to features of sclerosing cholangitis. The differential diagnostic considerations in visually apparent sclerosing cholangitis cover a diverse array of conditions apart from PSC.
Association of PSC with inflammatory bowel disease
A clinical hallmark of PSC is the high number of patients suffering from inflammatory bowel disease (IBD). In several studies with 605 PSC patients in the US (Mayo Clinic), UK (King’s College) and in Sweden, IBD was found in 71%, 73% and 81% of PSC cases (Boberg 1998, Bergquist 2002). In our own experience it is found in 52% of cases (Tischendorf 2007). Ulcerative colitis is more often associated (UK 71%, Sweden 72%) than Crohn’s disease. IBD is usually diagnosed before PSC but owing to the symptomatic latency of both IBD and PSC it can also be diagnosed at the same time or later than PSC. Most commonly ulcerative colitis is diagnosed more than a year before PSC (67%). This is backed by genome wide association data (Janse 2011). In 22% the diagnoses occurred within one year of each other, and only in 11% the diagnosis of ulcerative colitis reached more than one year after PSC was established. IBD patients with elevated liver biochemistry are a risk group and require careful hepatological workup for PSC. About 5% of all patients with ulcerative colitis have PSC.
PSC as a risk factor for cancer
Apart from the risk of developing portal hypertension and cirrhosis, PSC is a severe risk factor for cancer, which distinguishes this disease from AIH and PBC (Table 8). The increased risk of cholangiocarcinoma is well described (Bergquist 2001, Boberg 2002). The numbers reported vary because explanted livers during liver transplantation, autopsies and in vivo diagnosed cases are taken into account in different analyses. The diagnosis of cholangiocarcinoma (CC) in PSC patients continues to represent a difficult task because stenoses upon cholangiography may be caused by inflammatory activity as well as tumour, and because biochemical tests and biopsy procedures have a low sensitivity and specificity. Imaging studies are equally complicated by a lack of sensitivity since tumours frequently grow intramurally and are diagnosed in late stages precluding curative therapeutic approaches. Studies from Sweden show that 54% of CC occurs within one year of the diagnosis of PSC and 27% are diagnosed at liver transplantation. Overall, 12.2% of Northern European PSC patients develop CC, which is corroborated by our data from Hannover (Boberg 2002, Tischendorf 2006). These patients suffer from jaundice, pruritus and abdominal pains and had a longer IBD history. Male gender and smoking are also risk factors (Tischendorf 2006, Weismüller 2008). In a Dutch study there were similar findings of 18 CC out of 174 patients (10%) (Ponsioen 2002). The CC risk of a PSC patient amounts to 1.5% per year and is 161-fold higher than in healthy controls. In the future, the option of proteomic analyses of bile (and urine) may be of importance to predict the risk of cancer (Metzger 2013).
It is also important to realise that the risk for colorectal cancer (CRC) is elevated 10-fold, in addition to a 14-fold risk of pancreatic cancer (Bergquist 2002). These data point to the need of annual colonoscopies and ultrasound studies after diagnosis of PSC to monitor the high potential for cancer development.
Table 8. Cancer association of PSC| Cholangiocarcinoma | 10–20% of PSC patients Yearly risk 1.5% Frequent within 1 year of diagnosis Bilirubin, male gender, long-standing ulcerative colitis, abdominal symptoms, smoking |
| Colorectal cancer | 10-fold risk (PSC and ulcerative colitis) Yearly colonoscopies in ulcerative colitis In ulcerative colitis and AP elevation: consider ERC |
| Pancreatic cancer | 14-fold risk in PSC patients Abdominal ultrasound |
Medical therapy of PSC
Present day data and clinical experience do not suggest that PSC can be curable by medical therapy (Zein 2010, Wiencke 2011). A cure would include the improvement or normalisation of abnormal cholestatic biochemical features but more importantly the improvement of sclerosing changes to the intra- and extrahepatic biliary tree, which ultimately lead to biliary cholangitis, to episodes of cholangitis, and, which carry the risk of cholangiocellular carcinoma. The only available drug that combines a favourable toxicity profile and can lead to a reduction of cholestatic serum parameters currently is ursodeoxycholic acid (UDCA). However, controversy surrounds the use of UDCA (Chapman 2010), which has recently led to guidelines that do not specifically recommend UDCA treatment in all adult patients (Guidelines 2009, Chapman 2010).
In two studies an improvement was documented using 20 mg/kg body weight, and 25–30 mg/kg body weight, respectively (Harnois 2001, Mitchell 2001). Both use UDCA doses, which are considerably higher than those common in the therapy of PBC (15 mg/kg body weight). From these data a higher dose appeared to be more beneficial in PSC. However, a study analysing UDCA in bile as a function of oral UDCA dose found that doses exceeding 25 mg/kg body weight are not likely to be useful since the maximum transport of UDCA into the bile leveled off at 25 mg/kg with no further increase (Rost 2004). After these and other initial reports, a meta-analysis was published in 2002 (Chen 2003), that concluded that UDCA therapy improved biochemical parameters but that overall benefit in patients with PSC, in particular survival benefit, was uncertain. A large study appeared to confirm this: 219 PSC patients in a placebo-controlled trial (Olsson 2005) received 17 to 23 mg/kg body weight of UDCA and a trend towards better survival and less need for transplantation was seen, but did not reach statistical significance. A difference in the incidence of cholangiocarcinoma was not observed. However, statistical analyses reported in this study concluded that 346 patients would have been required to reach statistical significance.
Recent reports show that the withdrawal of UDCA – which would follow the US practice guidelines - leads to a biochemical deterioration in PSC patients (Wunsch 2014). As in PBC biochemical remission appears to be associated with a favourable prognosis also in PSC patients (Lindstrom 2013). Therefore, based on the body of literature available, a positive effect of UDCA at present cannot be excluded, withdrawal may not be in the best interest of the patient, and clearly larger placebo-controlled studies are required. This will only be possible in multicentre trials, which are not likely to be conducted in the near future.
The issue of a protective effect of UDCA on colonic neoplasia reported in the past has not been replicated (Lindstrom 2012).
The issue of immunosuppression in PSC is controversial and the majority of centres and publications do not recommend the routine administration of corticosteroids and other immunosuppressants (van Hoogstraten 2000, LaRusso 2006). In PSC one of the most feared and unpredictable complicating factors is bacterial cholangitis and cholangiosepsis (Negm 2011). Immunosuppression would be expected to aggravate this complication. In rare instances such as overlapping features of PSC and autoimmune hepatitis (AIH) (Boberg 2011), immunosuppression may be of benefit but this requires rigorous documentation of AIH, which includes biopsies, autoimmune serology and suggestive biochemistry (Boberg 1996, Beuers 2005).
A potential future drug is nor-UDCA, which is being evaluated in clinical studies. Nor-UDCA undergoes a different shunting route than UDCA and is not conjugated. In animal models nor-UDCA has led to significant effects on the development and progression of sclerosing cholangitis (Fickert 2006, Fickert 2013). Meanwhile a phase 2 trial has been completed and reported at international meetings but not yet published. Nor-UDCA was shown to reduce biochemical parameters in a dose-dependent fashion irrespective of prior treatment with UDCA or UDCA treatment failure. No safety issues were reported and a phase 3 study is planned.
Therapy of IBD in PSC
Many PSC patients suffer from a milder course of IBD. Ulcerative colitis is frequently characterised by pancolitis without severe symptoms, rectal sparing or backwash ileitis. Nevertheless the risk of dysplasia and CRC remains significantly higher in PSC patients with ulcerative colitis. Therapeutic intervention is no different that that for IBD without PSC.
Endoscopic therapy
The most important factor determining the course of PSC is the development of biliary strictures, which carry and increase the risk of septic cholangitis driving biliary fibrosis (Figures 4 and 5). Endoscopic dilatation can improve cholestasis, in some cases biliary stenting (Weismüller 2008), which is not recommended by all gastroenterologists. The international PSC study group is conducting a prospective study (DilStent Study) to evaluate stenting versus balloon dilatation therapy in PSC. The combination of endoscopic intervention and UDCA therapy appears to lead to a significant prolongation of transplant-free survival. UDCA alone does not lead to this effect.
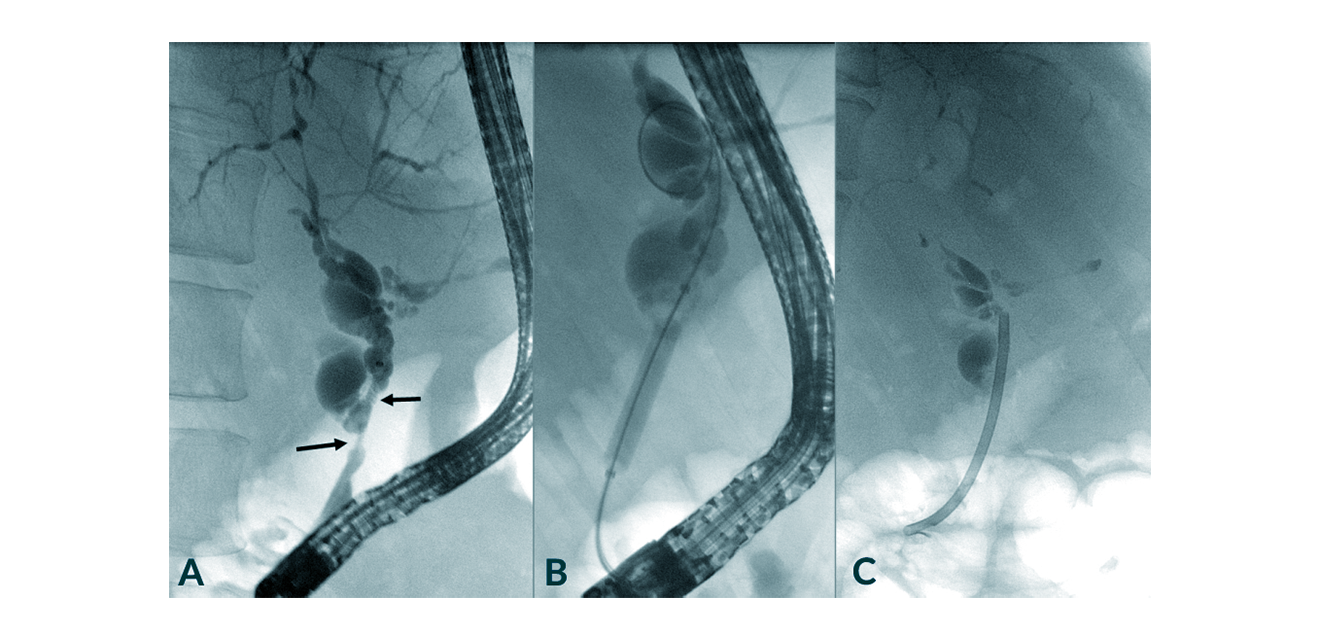 Figure 6. Management of PSC by dilatation of a dominant stricture of the common bile duct (arrows) and subsequent short-term stenting with a plastic stent. In this particular case it turned out that the biliary biopsy revealed cholangiocarcinoma
Figure 6. Management of PSC by dilatation of a dominant stricture of the common bile duct (arrows) and subsequent short-term stenting with a plastic stent. In this particular case it turned out that the biliary biopsy revealed cholangiocarcinoma
Liver transplantation in PSC (OLT)
In PSC patients survival has been shown to be reduced both in symptomatic and in asymptomatic patients (Kim 2000, LaRusso 2006), which is in part attributable to the inherent risk of cholangiocarcinoma affecting 10–20% of these patients, and renders decision-making for liver transplantation a formidable challenge. In addition, PSC patients with advanced destructive cholangiopathy frequently exhibit only mild signs of liver failure based upon coagulation abnormalities, hypoalbuminaemia, or complications of portal hypertension (Tischendorf 2007, Strassburg 2009). The course of deterioration to liver failure is often observed after long periods of clinical stability, and frequently proceeds rapidly following septic biliary complications. This is not well predicted by the aforementioned PSC scores, which is also true for the model of end-stage liver disease (MELD), the measure for organ allocation in the US and the Eurotransplant member countries.
Two major problems define the challenges involved in the indication for liver transplantation in PSC. Firstly, timing is difficult (Wiesner 1992). PSC patients are young and preemptive liver transplantation carries a higher short-term risk of OLT itself than the most likely short-term natural course of the disease. On the other hand, patients that urgently require OLT because of advanced biliary destruction frequently do not meet priority criteria calculated by the MELD system. In Germany, allocation can proceed by stand exception priority if PSC patients fulfill the requirements. Secondly, the 161-fold increase of cholangiocarcinoma risk (Bergquist 2002) is a risk that may eliminate the option of liver transplantation altogether when evidence of cholangiocarcinoma is detected by diagnostic imaging procedures. The diagnosis of early cholangiocarcinoma is difficult and presently no single diagnostic procedure with high sensitivity and specificity is available (Tischendorf 2006). Moreover, the patients at risk cannot be reliably identified.
In terms of practical management the first point can only be addressed by careful clinical monitoring of PSC patients in experienced hepatological transplant centres, where the likelihood of early complication diagnosis and management, as well as the individualised timing of wait-listing for OLT is higher (Tischendorf 2007). The second point has been addressed in two centres by establishing specific protocols for the management of hilar cholangiocarcinoma and OLT (Sudan 2002, Rea 2005). A rigorous algorithm for non-resectable hilar cholangiocarcinoma patients who were carefully selected and capable of surviving chemotherapy, radiation therapy and surgery was reported. A multimodal approach including neoadjuvant chemo-/radiation therapy, brachytherapy, chemotherapy, laparotomy and OLT was employed resulting in a five-year survival of 82%, which did not differ from results in PSC patients without cholangiocarcinoma (Rea 2005). However, although attractive, these interdisciplinary strategies are best limited to studies and experienced hepatological transplant centres.
Overall the results of liver transplantation in PSC are good, leading to 10-year survival rates of around 70% (Graziadei 1999). In our centre, the median survival of PSC patients with cholangiocarcinoma was 12.7 months, and all PSC patients, irrespective of OLT, had a mean survival of 112 months (Tischendorf 2006). Recurrence after OLT is difficult to diagnose but appears to occur in up to 25% of patients (Graziadei 1999, Rowe 2008). Liver transplantation continues to represent the only curative option in PSC. Future developments will have to address the low sensitivity and specificity of early cholangiocarcinoma detection, the clinical prediction of the course of disease and consequently, specific allocation criteria for patients with PSC.
References
Ahmed M, Mutimer D, Hathaway M, Hubscher S, McMaster P, Elias E. Liver transplantation for autoimmune hepatitis: a 12-year experience. Transplant Proc 1997;29:496.
Al-Benna S, Willert J, Steinau HU, Steinstraesser L. Secondary sclerosing cholangitis, following major burn injury. Burns 2011;36(6):e106-10.
Al-Chalabi T, Boccato S, Portmann BC, McFarlane IG, Heneghan MA. Autoimmune hepatitis (AIH) in the elderly: a systematic retrospective analysis of a large group of consecutive patients with definite AIH followed at a tertiary referral centre. J Hepatol 2006;45(4):575-83.
Al-Chalabi T, Portmann BC, Bernal W, McFarlane IG, Heneghan MA. Autoimmune hepatitis overlap syndromes: an evaluation of treatment response, long-term outcome and survival. Aliment Pharmacol Ther 2008;28(2):209-20.
Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatology 1999;31:929-38.
Alvarez F, Ciocca M, Canero-Velasco C, et al. Short-term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol 1999;30:222-7.
Angulo P, El-Amin O, Carpenter HA, Lindor KD. Development of autoimmune hepatitis in the setting of long-standing primary biliary cirrhosis. Am J Gastroenterol 2001;96(10):3021-7.
Angulo P, Smith C, Jorgensen RA. Budesonide in the treatment of patients with primary biliary cirrhosis with suboptimal response to ursodeoxycholic acid. Hepatology 2000;20:471-90.
Aqel BA, Machicao V, Rosser B, Satyanarayana R, Harnois DM, Dickson RC. Efficacy of tacrolimus in the treatment of steroid refractory autoimmune hepatitis. J Clin Gastroenterol 2004;38(9):805-9.
Bach N, Bodian C, Bodenheimer H, et al. Methotrexate therapy for primary biliary cirrhosis. Am J Gastroenterol 2003;98(1):187-93.
Ben-Ari Z, Dhillon AP, Sherlock S. Autoimmune cholangiopathy: part of the spectrum of autoimmune chronic active hepatitis. Hepatology 1993;18(1):10-5.
Bergquist A, Broome U. Hepatobiliary and extra-hepatic malignancies in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol 2001;15(4):643-56.
Bergquist A, Ekbom A, Olsson R, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36(3):321-7.
Beuers U, Rust C. Overlap syndromes. Semin Liver Dis 2005;25(3):311-20.
Beuers U, Gershwin ME, Gish RG, et al. Changing nomenclature for PBC: From ‘cirrhosis’ to ‘cholangitis’. J Hepatol. 2015;63(5):1285-7.
Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol 1998;33:99-103.
Boberg KM, Bergquist A, Mitchell S, et al. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand J Gastroenterol 2002;37(10):1205-11.
Boberg KM, Chapman RW, Hirschfield GM, Lohse AW, Manns MP, Schrumpf E. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011;54(2):374-85.
Boberg KM, Fausa O, Haaland T, et al. Features of autoimmune hepatitis in primary sclerosing cholangitis: an evaluation of 114 primary sclerosing cholangitis patients according to a scoring system for the diagnosis of autoimmune hepatitis. Hepatology 1996;23(6):1369-76.
Bodenheimer H, Jr., Schaffner F, Pezzullo J. Evaluation of colchicine therapy in primary biliary cirrhosis. Gastroenterology 1988;95(1):124-9.
Bodenheimer HC Jr, Schaffner F, Sternlieb I, et al. A prospective clinical trial of D-penicillamine in the treatment of primary biliary cirrhosis. Hepatology 1985;5(6):1139-42.
Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatology;56:1181-1188.
Bowlus CL. Obeticholic acid for the treatment of primary biliary cholangitis in adult patients: clinical utility and patient selection. Hepat Med. 2016;8:89-95.
Broome U, Glaumann H, Lindstom E, et al. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC). J Hepatol 2002;36(5):586-9.
Calmus Y, Gane P, Rouger P, Poupon R. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology 1990;11(1):12-5.
Campsen J, Zimmerman MA, Trotter JF, et al. Liver transplantation for autoimmune hepatitis and the success of aggressive corticosteroid withdrawal. Liver Transpl 2008;14(9):1281-6.
Cancado ELR, Porta G. Autoimmune hepatitis in South America. Dordrecht, Boston, London, Kluwer Academic Publishers, 2000.
Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51(2):660-78.
Chapman RW. Primary sclerosing cholangitis: what is the role of ursodeoxycholic acid in therapy for PSC? Nat Rev Gastroenterol Hepatol 2010;7(2):74-5.
Chazouilleres O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998;28(2):296-301.
Chazouilleres O, Wendum D, Serfaty L, Rosmorduc O, Poupon R. Long term outcome and response to therapy of primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. J Hepatol 2006;44(2):400-6.
Chen W, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev 2003(2):CD003626.
Christensen E, Neuberger J, Crowe J, et al. Beneficial effect of azathioprine and prediction of prognosis in primary biliary cirrhosis. Final results of an international trial. Gastroenterology 1985;89(5):1084-91.
Clendenon JN, Aranda-Michel J, Krishna M, Taner CB, Willingham DL. Recurrent liver failure caused by IgG4 associated cholangitis. Ann Hepatol 2011;10(4):562-4.
Colombato LA, Alvarez F, Cote J, Huet PM. Autoimmune cholangiopathy: the result of consecutive primary biliary cirrhosis and autoimmune hepatitis? Gastroenterology 1994;107(6):1839-43.
Corpechot C, Chazouillières O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long term aoutcome. J Hepatol 2011;55(6):1361-1367.
Czaja AJ, Carpenter HA, Lindor KD. Ursodeoxycholic acid as adjunctive therapy for problematic type 1 autoimmune hepatitis: a randomized placebo-controlled treatment trial. Hepatology 1999;30(6):1381-6.
Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. Significance of HLA DR4 in type 1 autoimmune hepatitis. Gastroenterology 1993;105:1502-7.
Czaja AJ, Carpenter HA. Distinctive clinical phenotype and treatment outcome of type 1 autoimmune hepatitis in the elderly. Hepatology 2006;43(3):532-8.
Czaja AJ, Lindor KD. Failure of budesonide in a pilot study of treatment-dependent autoimmune hepatitis. Gastroenterology 2000;119(5):1312-6.
Czaja AJ, Manns MP. Advances in the diagnosis, pathogenesis, and management of autoimmune hepatitis. Gastroenterology 2010;139(1):58-72 e4.
Danielson A, Prytz H. Oral budesonide for treatment of autoimmune chronic hepatitis. Aliment Pharmacol Ther 1994;8:585-90.
Debray D, Maggiore G, Giradet JP, Mallet E, Bernard O. Efficacy of cyclosporin A in children with type 2 autoimmune hepatits. J Pediatr 1999;135:111-4.
Devlin J, Donaldson P, Portman B, et al. Recurrence of autoimmune hepatitis following liver transplantation. Liver Transpl Surg 1995;1:162-5.
Dienes HP, Popper H, Manns M, Baumann W, Thoenes W, Meyer zum Büschenfelde K-H. Histologic features in autoimmune hepatitis. Z Gastroenterol 1989;27:327-30.
Donaldson PT. Immunogenetics in liver disease. Baillieres Clin Gastroenterol 1996;10(3):533-49.
Duclos-Vallee JC, Hadengue A, Ganne-Carrie N, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. Corticoresistance and effective treatment by cyclosporine A. Dig Dis Sci 1995;40(5):1069-73.
EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009;51(2):237-67.
EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol. 2015;63(4):971-1004.
Efe C, Purnak T, Ozaslan E. Anti TNF-alpha therapy can be a novel treatment option in patients with autoimmune hepatitis. Aliment Pharmacol Ther 2010;32(1):115; author reply 6-7.
Esposito I, Kubisova A, Stiehl A, Kulaksiz H, Schirmacher P. Secondary sclerosing cholangitis after intensive care unit treatment: clues to the histopathological differential diagnosis. Virchows Arch 2008;453(4):339-45.
Fernandez NF, Redeker AG, Vierling JM, Villamil FG, Fong T-L. Cyclosporine therapy in patients with steroid resistant autoimmune hepatitis. Am J Gastroenterol 1999;94:241-8.
Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2006;130(2):465-81.
Fickert P, Pollheimer MJ, Silbert D, et al. Differential effects of norUDCA and UDCA in obstructive cholestasis in mice. Journal of hepatology 2013;58(6):1201-8.
Floreani A, Sun Y, Zou ZS, et al. Proposed therapies in primary biliary cholangitis. Expert Rev Gastroenterol Hepatol. 2016;10(3):371-82.
Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl 2006;12(12):1813-24.
Geall MG, Schoenfield LJ, Summerskill WHJ. Classification and treatment of chronic active liver disease. Gastroenterology 1968;55:724-9.
Gelbmann CM, Rummele P, Wimmer M, et al. Ischemic-like cholangiopathy with secondary sclerosing cholangitis in critically ill patients. Am J Gastroenterol 2007;102(6):1221-9.
Gelpi C SE, Rodriguez-Sanchez JL. Autoantibodies against a serine tRNA-protein complex implicated in cotranslational selenocysteine insertion. Proc Natl Acad Sci U S A 1992;89(20):9739-43.
Gossard AA, Lindor KD. Development of autoimmune hepatitis in primary biliary cirrhosis. Liver Int 2007;27(8):1086-90.
Götz G, Neuhaus R, Bechstein WO, et al. Recurrence of autoimmune hepatitis after liver transplantation. Transplant Proc 1999;31:430-1.
Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic acid therapy for primary biliary cirrhosis: a meta analysis. Lancet 1999;354:1053-60.
Graziadei IW, Wiesner RH, Batts KP, et al. Recurrence of primary sclerosing cholangitis following liver transplantation. Hepatology 1999;29(4):1050-6.
Gregorio GV, Portman B, Reid F, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology 1997;25:541-7.
Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology 2001;33(3):544-53.
Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol 2001;96(5):1558-62.
Hendrickse MT, Rigney E, Giaffer MH, et al. Low-dose methotrexate is ineffective in primary biliary cirrhosis: long-term results of a placebo-controlled trial. Gastroenterology 1999;117(2):400-7.
Heneghan MA, McFarlane IG. Current and novel immunosuppressive therapy for autoimmune hepatitis. Hepatology 2002;35(1):7-13.
Hennes EM, Oo YH, Schramm C, et al. Mycophenolate mofetil as second line therapy in autoimmune hepatitis? Am J Gastroenterol 2008;103(12):3063-70.
Hennes EM, Zeniya M, Czaja AJ, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008;48(1):169-76.
Hirschfield GM, Mason A, Luketic V, et al. Efficacy of Obeticholic Acid in Patients with Primary Biliary Cirrhosis and Inadequate Response to Ursodeoxycholic Acid. Gastroenterology 2014;doi: 10.1053/j.gastro.2014.12.005.
Homberg JC, Abuaf N, Bernard O, et al. Chronic active hepatitis associated with anti liver/kidney microsome type 1: a second type of “autoimmune” hepatitis. Hepatology 1987;7:1333-9.
Ikeda T, Tozuka S, Noguchi O. Effects of additional administration of colchicine in ursodeoxycholic acid-treated patients with primary biliary cirrhosis: a prospective randomized study. J Hepatol 1996;24:88-94.
Janse M, Lamberts LE, Franke L, et al. Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL, and CARD9. Hepatology 2011;53(6):1977-85.
Jepsen P, Gronbaek L, Vilstrup H. Worldwide Incidence of Autoimmune Liver Disease. Dig Dis. 2015;33 Suppl 2:2-12.
Johnson PJ, McFarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N EnglJ Med 1995;333:958-63.
Johnson PJ, McFarlane IG. Meeting report: International autoimmune hepatitis group. Hepatology 1993;18:998-1005.
Jones DE, James OF, Portmann B, Burt AD, Williams R, Hudson M. Development of autoimmune hepatitis following liver transplantation for primary biliary cirrhosis. Hepatology 1999;30(1):53-7.
Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol 1999;30(3):402-7.
Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet 2014; 10.1016/s0140-6736(14)60720-0.
Kanzler S, Gerken G, Dienes HP, Meyer zum Büschenfelde K-H, Lohse AW. Cyclophosphamide as alternative immunosuppressive therapy for autoimmune hepatitis - report of three cases. Z Gastroenterol 1996;35:571-8.
Kaplan MM, Alling DW, Zimmerman HJ, et al. A prospective trial of colchicine for primary biliary cirrhosis. N Engl J Med 1986;315(23):1448-54.
Kato Y, Suzuki K, Kumagai M, et al. Familial primary biliary cirrhosis. Immunological and genetic study. Am J Gastroenterol 1981;75(3):188-91.
Kerkar N, Hadzic N, Davies ET, et al. De-novo autoimmune hepatitis after liver transplantation. Lancet 1998;351(9100):409-13.
Kim WR, Therneau TM, Wiesner RH, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000;75(7):688-94.
Kirk AP, Jain S, Pocock S, Thomas HC, Sherlock S. Late results of the Royal Free Hospital prospective controlled trial of prednisolone therapy in hepatitis B surface antigen negative chronic active hepatitis. Gut 1980;21:7893.
Kuiper EM, Hanse BE, De Vries RA, et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology 2009;136:1281-1287.
LaRusso NF, Shneider BL, Black D, et al. Primary sclerosing cholangitis: summary of a workshop. Hepatology 2006;44(3):746-64.
Lenzen H, Weismuller TJ, Negm AA, et al. Antineutrophil cytoplasmic antibodies in bile are associated with disease activity in primary sclerosing cholangitis. Scandinavian journal of gastroenterology 2013; 48(10): 1205-12.
Leuschner M, Guldutuna S, You T, Hubner K, Bhatti S, Leuschner U. Ursodeoxycholic acid and prednisolone versus ursodeoxycholic acid and placebo in the treatment of early stages of primary biliary cirrhosis. J Hepatol 1996;25(1):49-57.
Leuschner M, Holtmeier J, Ackermann H, et al. The influence of sulindac on patients with primary biliary cirrhosis that responds incompletely to ursodeoxycholic acid: a pilot study. Eur J Gastroenterol Hepatol 2002;14(12):1369-76.
Leuschner M, Maier KP, Schlichting J, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology 1999;117(4):918-25.
Lindstrom L, Boberg KM, Wikman O, et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Alimentary pharmacology & therapeutics 2012;35(4):451-7.
Lindstrom L, Hultcrantz R, Boberg KM, Friis-Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 2013;11(7):841-6.
Liu JZ, Hov JR, Folseraas T, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature genetics 2013;45(6):670-5.
Lombard M, Portmann B, Neuberger J, et al. Cyclosporin A treatment in primary biliary cirrhosis: results of a long-term placebo controlled trial. Gastroenterology 1993;104(2):519-26.
Mackay IR, Taft LI, Cowling DC. Lupoid hepatitis. Lancet 1956;2:1323-6.
Mackay IR, Gershwin ME. The nature of autoimmune disease. Semin Liver Dis 1997;17(1):3-11.
Manns M, Gerken G, Kyriatsoulis A, Staritz M, Meyer zum Büschenfelde KH. Characterisation of a new subgroup of autoimmune chronic active hepatitis by autoantibodies against a soluble liver antigen. Lancet 1987;1(8528):292-4.
Manns MP, Bahr MJ. Recurrent autoimmune hepatitis after liver transplantation - when non-self becomes self. Hepatology 2000;32(4):868-70.
Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and management of autoimmune hepatitis. Hepatology 2010;51(6):2193-213.
Manns MP, Griffin KJ, Sullivan KF, Johnson EF. LKM-1 autoantibodies recognize a short linear sequence in P450IID6, a cytochrome P-450 monooxygenase. J Clin Invest 1991;88:1370-8.
Manns MP, Johnson EF, Griffin KJ, Tan EM, Sullivan KF. Major antigen of liver kidney microsomal antibodies in idiopathic autoimmune hepatitis is cytochrome P450db1. J Clin Invest 1989;83:1066-72.
Manns MP, Kruger M. Immunogenetics of chronic liver diseases. Gastroenterology 1994;106(6):1676-97.
Manns MP, Strassburg CP. Autoimmune hepatitis: clinical challenges. Gastroenterology 2001;120(6):1502-17.
Manns MP, Woynarowski M, Kreisel W, et al. Budesonide induces remission more effectively than prednisone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology 2010;139(4):1198-206.
Metzger J, Negm AA, Plentz RR, et al. Urine proteomic analysis differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Gut 2013;62(1):122-130.
Michieletti P, Wanless IR, Katz A, et al. Antimitochondrial antibody negative primary biliary cirrhosis: a distinct syndrome of autoimmune cholangitis. Gut 1994;35(2):260-5.
Milkiewicz P, Hubscher SG, Skiba G, Hathaway M, Elias E. Recurrence of autoimmune hepatitis after liver transplantation. Transplantation 1999;68(2):253-6.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001;121(4):900-7.
Mitchison HC, Palmer JM, Bassendine MF, Watson AJ, Record CO, James OF. A controlled trial of prednisolone treatment in primary biliary cirrhosis. Three-year results. J Hepatol 1992;15(3):336-44.
Nakamura K, Yoneda M, Yokohama S, et al. Efficacy of ursodeoxycholic acid in Japanese patients with type 1 autoimmune hepatitis [see comments]. J Gastroenterol Hepatol 1998;13(5):490-5.
Negm AA, Schott A, Vonberg RP, et al. Routine bile collection for microbiological analysis during cholangiography and its impact on the management of cholangitis. Gastrointest Endosc 2011;72(2):284-91.
Neuberger J, Portmann B, Calne R, Williams R. Recurrence of autoimmune chronic active hepatitis following orthotopic liver grafting. Transplantation 1984;37(4):363-5.
Nevens F, Andreone P, Mazzella G, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med. 2016;375(7):631-43.
Newton JL, Burt AD, Park JB, Mathew J, Bassendine MF, James OF. Autoimmune hepatitis in older patients. Age Ageing 1997;26(6):441-4.
Nikias GA, Batts KP, Czaja AJ. The nature and prognostic implications of autoimmune hepatitis with acute presentation. J Hepatol 1994;21:866-71.
Nishioka M, Morshed SA, Kono K, et al. Frequency and significance of antibodies to P450IID6 protein in Japanese patients with chronic hepatiis C. J Hepatol 1997;26:992-1000.
Nishioka M, Morshed SA, McFarlane IG. Geographical variation in the frequency and characteristics of autoimmune liver diseases. Amsterdam: Elsevier; 1998.
Notghi A, Nestle U, Rittner G, et al. Chromosomal aberrations in patients with primary biliary cirrhosis. Hum Genet 1990;85(5):546-50.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology 2005;129(5):1464-72.
Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36(3):525-31.
Ponsioen CY, Vrouenraets SM, Prawirodirdjo W, et al. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a Dutch population. Gut 2002;51(4):562-6.
Potthoff A, Deterding K, Trautwein C, et al. Steroid treatment for severe acute cryptogenic hepatitis. Z Gastroenterol 2007;45(1):15-9.
Poupon R, Chazouilleres O, Corpechot C, Chretien Y. Development of autoimmune hepatitis in patients with typical primary biliary cirrhosis. Hepatology 2006;44(1):85-90.
Poupon RE, Huet PM, Poupon R, Bonnand AM, Nhieu JT, Zafrani ES. A randomized trial comparing colchicine and ursodeoxycholic acid combination to ursodeoxycholic acid in primary biliary cirrhosis. UDCA-PBC Study Group. Hepatology 1996;24(5):1098-103.
Poupon RE, Lindor KD, Cauch-Dudek K. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997;113:884-90.
Poupon RE, Lindor KD, Pares A, Chazouilleres O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol 2003;39(1):12-6.
Prados E, Cuervas-Mons V, De La Mata M, et al. Outcome of autoimmune hepatitis after liver transplantation. Transplantation 1998;66:1645-50.
Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev 2006;5(2):136-9.
Raedsch R, Stiehl A, Walker S, Scherrmann JM, Kommerell B. Combined ursodeoxycholic acid plus colchicine-treatment of primary biliary cirrhosis: results of a placebo-controlled double-blind study. Z Gastroenterol 1992;30 Suppl 1:55-7.
Ramos-Casals M, Brito-Zeron P, Soto MJ, Cuadrado MJ, Khamashta MA. Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Clin Rheumatol 2008;22(5):847-61.
Ratziu V, Samuel D, Sebagh M, et al. Long-term follow-up after liver transplantation for autoimmune hepatitis: evidence of recurrence of primary disease. J Hepatol 1999;30(1):131-41.
Rea DJ, Heimbach JK, Rosen CB, et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann Surg 2005;242(3):451-8; discussion 8-61.
Rebollo Bernardez J, Cifuentes Mimoso C, Pinar Moreno A, et al. Deflazacort for long-term maintenance of remission in type I autoimmune hepatitis. Rev Esp Enferm Dig 1999;91(9):630-8.
Richardson PD, James PD, Ryder SD. Mycophenolate mofetil for maintenance of remission in autoimmune hepatitis in patients resistant to or intolerant of azathioprine. J Hepatol 2000;33(3):371-5.
Rost D, Rudolph G, Kloeters-Plachky P, Stiehl A. Effect of high-dose ursodeoxycholic acid on its biliary enrichment in primary sclerosing cholangitis. Hepatology 2004;40(3):693-8.
Rowe IA, Webb K, Gunson BK, Mehta N, Haque S, Neuberger J. The impact of disease recurrence on graft survival following liver transplantation: a single centre experience. Transpl Int 2008;21(5):459-65.
Rust C, Beuers U. Overlap syndromes among autoimmune liver diseases. World J Gastroenterol 2008;14(21):3368-73.
Salcedo M, Vaquero J, Banares R, et al. Response to steroids in de novo autoimmune hepatitis after liver transplantation. Hepatology 2002;35(2):349-56.
Sanchez-Urdazpal L, Czaja AJ, Van Holk B. Prognostic features and role of liver transplantation in severe corticoid-treated autoimmune chronic active hepatitis. Hepatology 1991;15:215-21.
Schramm C, Kanzler S, zum Büschenfelde KH, Galle PR, Lohse AW. Autoimmune hepatitis in the elderly. Am J Gastroenterol 2001;96(5):1587-91.
Schreuder TC, Hubscher SG, Neuberger J. Autoimmune liver diseases and recurrence after orthotopic liver transplantation: what have we learned so far? Transpl Int 2009;22(2):144-52.
Silveira MG, Talwalkar JA, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary biliary cirrhosis: long-term outcomes. Am J Gastroenterol 2007;102(6):1244-50.
Silveira MG, Lindor KD. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert opinion on pharmacotherapy 2014;15(3):365-72.
Soloway RD, Summerskill WH, Baggenstoss AH, et al. Clinical, biochemical, and histological remission of severe chronic active liver disease: a controlled study of treatments and early prognosis. Gastroenterology 1972;63(5):820-33.
Stechemesser E, Klein R, Berg PA. Characterization and clinical relevance of liver-pancreas antibodies in autoimmune hepatitis. Hepatology 1993;18:1-9.
Strassburg CP, Bahr MJ, Becker T, Klempnauer J, Manns MP. Progress in immunosuppression. Chirurg 2008;79(2):149-56.
Strassburg CP, Becker T, Klempnauer J, Manns MP. Liver transplantation: deciding between need and donor allocation. Internist (Berl) 2004;45(11):1233-45.
Strassburg CP, Jaeckel E, Manns MP. Anti-mitochondrial antibodies and other immunological tests in primary biliary cirrhosis. Eur J Gastroenterol Hepatol 1999;11(6):595-601.
Strassburg CP, Manns MP. Liver transplantation: indications and results. Internist (Berl) 2009.
Strassburg CP, Manns MP. Primary biliary liver cirrhosis and overlap syndrome. Diagnosis and therapy. Internist (Berl) 2004;45(1):16-26.
Strassburg CP, Manns MP. Autoantibodies and autoantigens in autoimmune hepatitis. Semin Liver Dis 2002;22(4):339-52.
Strassburg CP, Manns MP. Autoimmune hepatitis in the elderly: what is the difference? J Hepatol 2006;45(4):480-2.
Strassburg CP, Manns MP. Autoimmune hepatitis versus viral hepatitis C. Liver 1995;15:225-32.
Strassburg CP, Manns MP. Primär biliäre Zirrhose und primär sklerosierende Cholangitis. Internistische Praxis 1996;36:57-74.
Strassburg CP, Obermayer-Straub P, Alex B, et al. Autoantibodies against glucuronosyltransferases differ between viral hepatitis and autoimmune hepatitis. Gastroenterology 1996;111(6):1576-86.
Strassburg CP, Obermayer-Straub P, Manns MP. Autoimmunity in liver diseases. Clin Rev Allergy Immunol 2000;18(2):127-39.
Strassburg CP. Autoimmune liver diseases and their overlap syndromes. Praxis (Bern 1994) 2006;95:1363-81.
Stravitz RT, Lefkowitch JH, Fontana RJ, et al. Autoimmune acute liver failure: proposed clinical and histological criteria. Hepatology 2011;53(2):517-26.
Sudan D, DeRoover A, Chinnakotla S, et al. Radiochemotherapy and transplantation allow long-term survival for nonresectable hilar cholangiocarcinoma. Am J Transplant 2002;2(8):774-9.
Takuma K, Kamisawa T, Igarashi Y. Autoimmune pancreatitis and IgG4-related sclerosing cholangitis. Curr Opin Rheumatol 2011;23(1):80-7.
Talor E, Rose NR. Hypothesis: the aging paradox and autoimmune disease. Autoimmunity 1991;8(3):245-9.
Terjung B, Spengler U, Sauerbruch T, Worman HJ. “Atypical p-ANCA” in IBD and hepatobiliary disorders react with a 50-kilodalton nuclear envelope protein of neutrophils and myeloid cell lines. Gastroenterology 2000;119(2):310-22.
Thalen A, Brattsand R. Synthesis and anti-inflammatory properties of budesonide, a new non-halogenated glucocorticoid with high local activity. Arzneimittelforschung 1979;29(11):1687-90.
Than NN, Wiegard C, Weiler-Normann C, et al. Long-term follow-up of patients with difficult to treat type 1 autoimmune hepatitis on Tacrolimus therapy. Scand J Gastroenterol. 2016;51(3):329-36.
Tillmann HL, Jackel E, Manns MP. Liver transplantation in autoimmune liver disease-selection of patients. Hepatogastroenterology 1999;46(30):3053-9.
Tischendorf JJ, Hecker H, Kruger M, Manns MP, Meier PN. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: A single center study. Am J Gastroenterol 2007;102(1):107-14.
Tischendorf JJ, Meier PN, Strassburg CP, et al. Characterization and clinical course of hepatobiliary carcinoma in patients with primary sclerosing cholangitis. Scand J Gastroenterol 2006;41(10):1227-34.
Tsuji K, Watanabe Y, Van De Water J, et al. Familial primary biliary cirrhosis in Hiroshima. J Autoimmun 1999;13(1):171-8.
Uibo R, Salupere V. The epidemiology of primary biliary cirrhosis: immunological problems. Hepatogastroenterology 1999;46(30):3048-52.
Umekita K, Miyauchi S, Ueno S, et al. Improvement of rheumatoid arthritis and autoimmune hepatitis in a patient treated with the tumour necrosis factor inhibitor, etanercept. Intern Med 2011;50(11):1245-9.
Van Gerven NM, van der Eijk AA, Pas SD, et al. Seroprevalence of Hepatitis E Virus in Autoimmune Hepatitis Patients in the Netherlands. J Gastrointestin Liver Dis. 2016;25(1):9-13.
Van Hoogstraten HJ, Vleggaar FP, Boland GJ, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double-blind pilot study. Belgian-Dutch PSC Study Group [see comments]. Am J Gastroenterol 2000;95(8):2015-22.
Van Steenbergen W, Sciot R, Van Eyken P, Desmet V, Fevery J. Combined treatment with methotrexate and ursodeoxycholic acid in non-cirrhotic primary biliary cirrhosis. Acta Clin Belg 1996;51(1):8-18.
Van Thiel DH, Wright H, Carroll P, et al. Tacrolimus: A potential new treatment for autoimmune chronic active hepatitis: results of an open-label preliminary trial. Am J Gastroenterol 1995;90:771-6.
Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014;59(6):2286-98.
Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453.
Vogel A, Heinrich E, Bahr MJ, et al. Long-term outcome of liver transplantation for autoimmune hepatitis. Clin Transplant 2004;18(1):62-9.
Volkmann M, Luithle D, Zentgraf H, et al. SLA/LP/tRNP((Ser)Sec) antigen in autoimmune hepatitis: identification of the native protein in human hepatic cell extract. J Autoimmun 2010;34(1):59-65.
Volkmann M, Martin L, Baurle A, et al. Soluble liver antigen: isolation of a 35-kd recombinant protein (SLA-p35) specifically recognizing sera from patients with autoimmune hepatitis. Hepatology 2001;33(3):591-6.
Von Figura G, Stephani J, Wagner M, et al. Secondary sclerosing cholangitis after chemotherapy with bevacizumab and paclitaxel. Endoscopy 2009;41 Suppl 2:E153-4.
Waldenström J. Leber, Blutproteine und Nahrungseiweisse. Dtsch Gesellsch Verd Stoffw 1950;15:113-9.
Warnes TW, Smith A, Lee FI, Haboubi NY, Johnson PJ, Hunt L. A controlled trial of colchicine in primary biliary cirrhosis. Trial design and preliminary report. J Hepatol 1987;5(1):1-7.
Webster GJ, Pereira SP, Chapman RW. Autoimmune pancreatitis/IgG4-associated cholangitis and primary sclerosing cholangitis – overlapping or separate diseases? J Hepatol 2009;51(2):398-402.
Weiler-Normann C, Schramm C, Quaas A, et al. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J Hepatol. 2013;58(3):529-34.
Weismüller TJ, Wedemeyer J, Kubicka S, Strassburg CP, Manns MP. The challenges in primary sclerosing cholangitis - Aetiopathogenesis, autoimmunity, management and malignancy. J Hepatol 2008;48(S1):S38-S57.
Wiegand J, Schuler A, Kanzler S, et al. Budesonide in previously untreated autoimmune hepatitis. Liver Int 2005;25(5):927-34.
Wiencke K, Boberg KM. Current consensus on the management of primary sclerosing cholangitis. Clin Res Hepatol Gastroenterol 2011;35(12):786-91.
Wies I, Brunner S, Henninger J, et al. Identification of target antigen for SLA/LP autoantibodies in autoimmune hepatitis [see comments]. Lancet 2000;355(9214):1510-5.
Wiesner RH, Ludwig J, Lindor KD, et al. A controlled trial of cyclosporine in the treatment of primary biliary cirrhosis. N Engl J Med 1990;322(20):1419-24.
Wiesner RH, Porayko MK, Dickson ER, et al. Selection and timing of liver transplantation in primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 1992;16(5):1290-9.
Wolfhagen FHJ, van Hoogstraaten HJF, van Buuren, H.R. Triple therapy with ursodeoxycholic acid, prednisone, and azathioprine in primary biliary cirrhosis: a 1 year randomized, placebo controlled study. J Hepatol 1998;29:736-42.
Woynarowski M, Nemeth A, Baruch Y, et al. Budesonide versus prednisone with azathioprine for the treatment of autoimmune hepatitis in children and adolescents. The Journal of paediatrics 2013;163(5):1347-53.e1.
Wright HL, Bou-Abboud CF, Hassanenstein T, et al. Disease recurrence and rejection following liver transplantation for autoimmune chronic active liver disease. Transplantation 1992;53:136-9.
Wunsch E, Trottier J, Milkiewicz M, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014;60(3):931-40.
Ytting H, Larsen FS. Everolimus treatment for patients with autoimmune hepatitis and poor response to standard therapy and drug alternatives in use. Scand J Gastroenterol. 2015;50(8):1025-31.
Zachou K, Gatselis NK, Arvaniti P, et al. A real-world study focused on the long-term efficacy of mycophenolate mofetil as first-line treatment of autoimmune hepatitis. Aliment Pharmacol Ther. 2016;43(10):1035-47.
Zein CO, Lindor KD. Latest and emerging therapies for primary biliary cirrhosis and primary sclerosing cholangitis. Curr Gastroenterol Rep 2010;12(1):13-22.
Zenouzi R, Weismuller TJ, Hubener P, et al. Low risk of hepatocellular carcinoma in patients with primary sclerosing cholangitis with cirrhosis. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association 2014;12(10):1733-8.
Zhang L, Lewis JT, Abraham SC, et al. IgG4+ plasma cell infiltrates in liver explants with primary sclerosing cholangitis. Am J Surg Pathol 2011;34:88-94.