
23. Wilson’s disease
Claus Niederau
Introduction
In 1912, Kinnear Wilson was the first to describe an inherited lethal disease associated with progressive lenticular degeneration, chronic liver disease and cirrhosis (Wilson 1912). In the same year, Kayser and Fleischer detected that patients with Wilson’s Disease (WD) often have brownish corneal copper deposits now called Kayser-Fleischer rings (Fleischer 1912).
WD is an autosomal recessive error of the metabolism. Its gene ATP7B encodes a copper-transporting ATPase (Bull 1993, Tanzi 1993, Petrukhin 1993, Yamaguchi 1993). The genetic defect of the ATP7B protein reduces biliary copper excretion leading to copper accumulation in the cornea and various organs including the liver, brain and kidney. The alteration of the ATP7B protein also reduces the incorporation of copper into ceruloplasmin. The corresponding presence of apoceruloplasmin (ceruloplasmin with no copper incorporation) leads to a decrease in circulating levels of ceruloplasmin due to the reduced half-life of the apoprotein. Thus, despite copper accumulation in many organs, circulating levels of copper and ceruloplasmin are decreased in most WD patients.
The prevalence of WD is rare, estimated at 3 per 100,000 in the general population (Frysman 1990). The clinical presentation may vary. Some WD patients are diagnosed with liver problems while others present with neurologic or psychiatric symptoms; many patients show both hepatic and neurological disease (Figure 1). Episodes of hemolysis and renal abnormalities may also occur. WD typically affects children and younger adults, and is rarely seen in adults older than 40. WD is fatal unless appropriately treated. Drugs for treatment of WD are copper chelators such as penicillamine, and trientine (Walshe 1956). More recently, zinc has been used to reduce intestinal copper absorption and to detoxify free circulating copper. Patients with fulminant liver failure or decompensated cirrhosis may have to undergo liver transplantation (LTX), which cures WD.
Clinical presentation
Screening for WD is only useful in families with an affected member. In all other circumstances diagnostic procedures are only done when symptoms and findings suggest WD. These include liver disease, neurological symptoms, renal abnormalities and episodes of hemolysis. WD is diagnosed in the vast majority of patients between the ages of 5 and 35. There are rare reports of patients diagnosed at ages 3–5 (Kalach 1993, Wilson 2000) and at ages of up to about 60 years (Gow 2000). Late-onset WD is a frequently overlooked condition (Ferenci 2007). Diagnostic workup does not rely on a single test but includes identification of corneal Kayser-Fleischer rings, reduced serum ceruloplasmin and copper as well as a quantitative determination of liver copper concentration (Scheinberg 1952, Walshe 1956, Saito 1987, Stremmel 1991, Roberts 2003) (Figure 2).
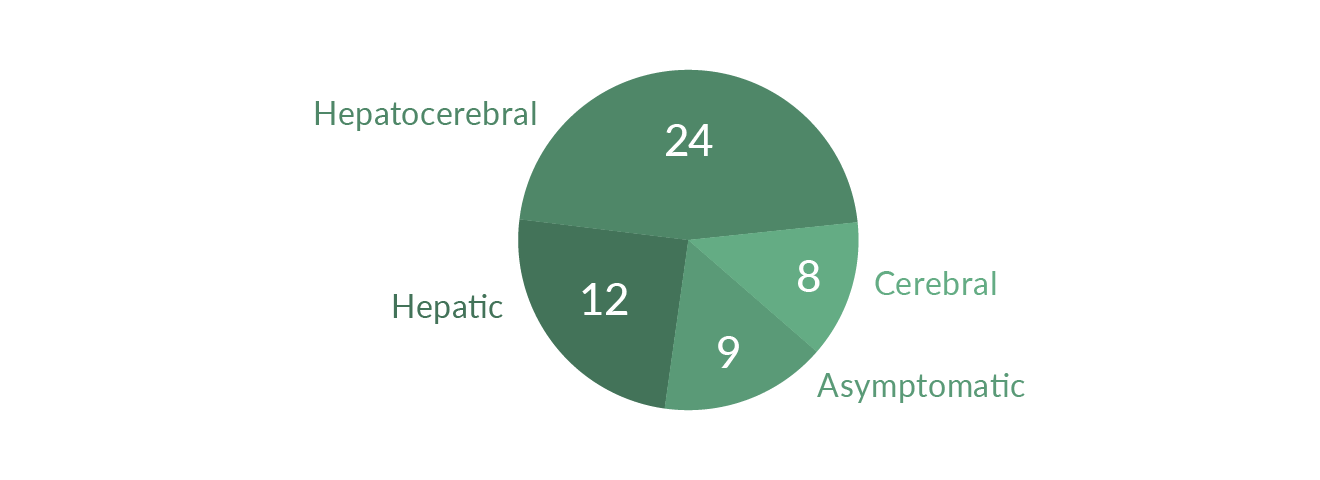 Figure 1. Clinical course of WD in 53 patients (modified from Stremmel 1991)
Figure 1. Clinical course of WD in 53 patients (modified from Stremmel 1991)
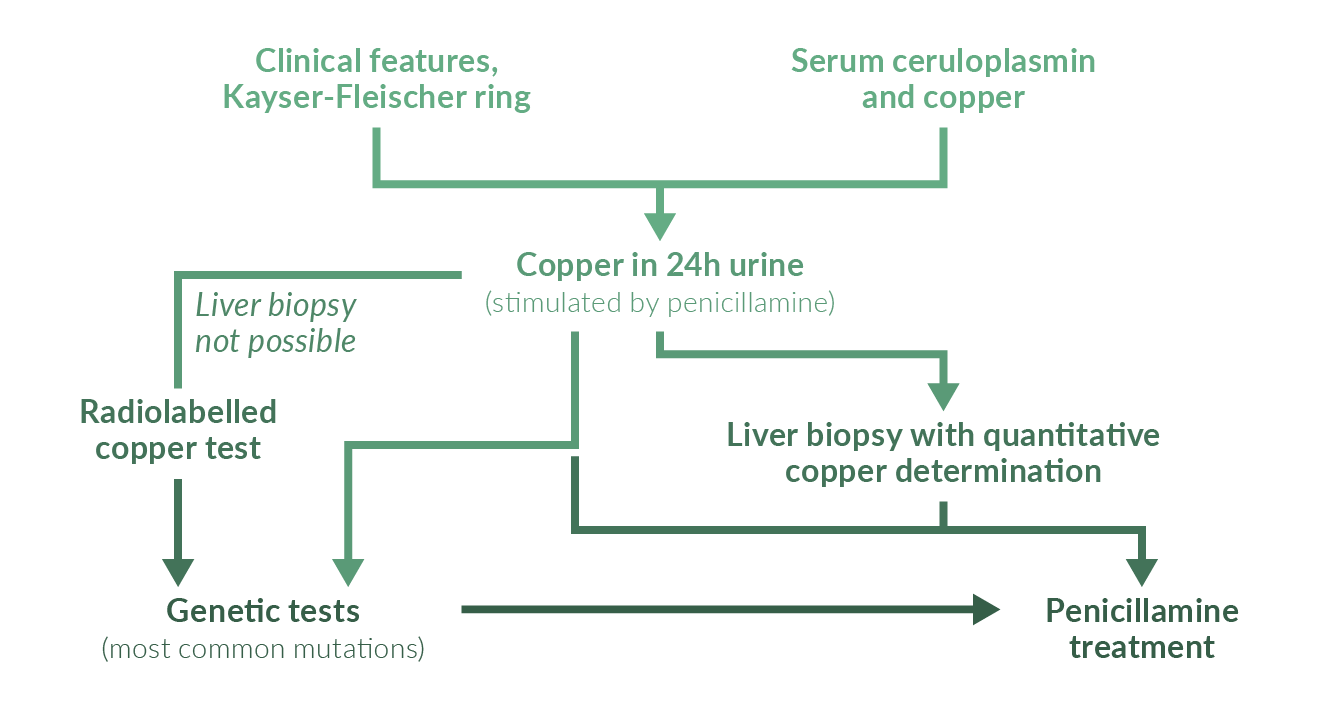 Figure 2. Diagnostic workup for WD
Figure 2. Diagnostic workup for WD
Genetic tests are usually only done in relatives of a confirmed WD patient. It is easy to diagnose WD in subjects who present with liver cirrhosis, typical neurologic manifestations and Kayser-Fleischer rings; many of these patients present at ages 5 to 35 and have decreased serum copper and ceruloplasmin (Sternlieb 1990). However, a considerable number of WD patients present only with liver disease and may not have Kayser-Fleischer rings or decreased serum levels of ceruloplasmin (Steindl 1997). Under these circumstances diagnosis may be difficult; measurement of 24-hour urinary copper excretion often helps to support the suspicion of WD. Liver biopsy with measurement of quantitative copper concentration should be done to corroborate the diagnosis (Stremmel 1991, Roberts 2003).
In general, WD patients diagnosed primarily with liver disease are children and adolescents and are younger than those diagnosed due to neurological symptoms (Merle 2007). Many patients who present only with CNS symptoms are 20–40 years old. Patients with WD may present with a wide spectrum of liver disease ranging from asymptomatic elevation of serum aminotransferases to fulminant liver failure. Serum aminotransferases are elevated in most WD patients irrespective of age (Schilsky 1991). Other WD patients may present with findings and symptoms of autoimmune hepatitis including autoimmune antibodies and elevated IgG (Scott 1978, Milkiewicz 2000). The clinical picture might also resemble acute or chronic viral hepatitis, without the viral serum markers. Even liver histology is not predictive or typical for WD unless copper concentration is measured. Histological findings may range from fatty liver changes to severe necro-inflammatory and fibrotic disease and complete cirrhosis. In particular, children and adolescents with chronic active hepatitis of unknown aetiology or autoimmune hepatitis and adult patients with a suspicion of autoimmune hepatitis or non-response to immunosuppressants should be evaluated for WD (Roberts 2003).
WD has to be excluded in patients with fulminant liver failure of unknown aetiology, especially at ages under 35 years; WD patients with such presentation usually have some sort of liver disease (Rector 1984, Ferlan-Maroult 1999, Roberts 2003) associated with Coombs negative hemolytic anaemia and severely increased prothrombine time non-responsive to vitamin K and progressive renal failure (Sallie 1992). Some patients have bilirubin levels of more than 40 mg/dL while serum alkaline phosphatase is normal or just slightly elevated (Berman 1991). In contrast to many types of toxic liver failure, liver failure in WD usually does not start with high increases in aminotransferases. In many WD patients AST levels exceed ALT levels (Emre 2001, Berman 1991). In most cohorts, for unexplained reasons, the ratio of females to males is approximately 2:1 (Roberts 2003). Serum ceruloplasmin may be decreased while serum copper and 24-hour urinary excretion of copper is usually elevated. It is extremely helpful when one can identify Kayser-Fleischer rings in this situation; these patients need to be studied with a slit lamp by an experienced ophthalmologist. Patients with acute liver failure need a diagnostic workup as rapidly as possible; if there is a strong suspicion or diagnosis of WD, the patient should be transferred to a transplant centre the same day.
Neurological symptoms in WD often resemble those seen in Parkinson’s disease including tremor and rigor. Many patients report that symptoms start with problems in handwriting and dysarthria. Neurological symptoms may be associated with slight behavioural alterations, which may later proceed to manifest psychiatric disease including depression, anxiety and psychosis. With the progression of CNS involvement WD patients may develop seizures and pseudobulbar palsy associated with severe dysphagia, aspiration and pneumonia. Although many older WD patients present with neurological disease, the diagnostic workup often shows significant liver involvement or even complete liver cirrhosis.
Renal involvement of WD may present with aminoaciduria and nephrolithiasis (Azizi 1989, Nakada 1994, Cu 1996). There may be various other non-neurological and non-hepatic complications of WD such as osteoporosis and arthritis, cardiomyopathy, pancreatitis, hypoparathyroidism, and miscarriages (for literature see Roberts 2003).
Kayser-Fleischer rings are caused by corneal copper deposition (Figure 3). Sometimes, one can see the rings directly as a band of brown pigment close to the limbus. In other patients the ring can only be identified using a slit lamp. Very rarely similar rings may be seen in non-WD patients, e.g., in some patients with neonatal or chronic cholestasis (Tauber 1993). Kayser-Fleischer rings are detectable in 50–60% of WD patients in most large cohorts (Tauber 1993, Roberts 2003). Many young WD patients with liver disease do not have such rings (Giacchino 1997) whereas almost all patients with primarily neurologic symptoms do have them (Steindl 1997). WD patients may also have other less specific eye changes including sunflower cataracts (Cairns 1963). Kayser-Fleischer rings usually regress with chelation therapy or after LTX (Stremmel 1991, Schilsky 1994).
 Figure 3. Kayser-Fleischer ring in a patient with WD
Figure 3. Kayser-Fleischer ring in a patient with WD
Diagnosis
Scoring system
Diagnosis of Wilson’s disease may be difficult. Therefore, a scoring system has been established (Ferenci 2003) (Table 1) which is now recommended by recent EASL guidelines (EASL 2012) (Table 2).
Table 1. Scoring system – 8th International Meeting on Wilson’s disease (Ferenci 2003)| KF rings | Absent 0 | Present 2 | |
| Neurologic symptoms | Absent =0 | Mild =1 | Severe =2 |
| Serum ceruloplasmin | >0.2 g/L =0 | 0.1–0.2 g/L =1 | <0.1 g/L =2 |
| Coombs negative hemolytic anaemia | Absent =0 | Present=1 | |
| Liver copper (in the absence of cholestasis) | 0.8 μmol/g =1 | 0.8–4 μmol/g =1 | >4 μmol/g =2 |
| Rhodanine positive granules | Absent =0 | Present =1 | |
| Urinary copper (in absence of acute hepatitis) | Normal =0 Normal =0 | 1–2x ULN =1 Normal, but >5xULN | >2x ULN =2 after D-penicillamine =2 |
| Mutation analysis | None =0 | 1 chromosome =1 | both chromosomes =4 |
| Total sore | |
| ≥4: | Diagnosis established |
| 3: | Diagnosis possible, more tests needed |
| 2: | Diagnosis very unlikely |
Serum ceruloplasmin
Ceruloplasmin, the major circulating copper transporter, is synthesised and secreted mainly by hepatocytes. The 132 kd protein consists of six copper atoms per molecule of ceruloplasmin (holoceruloplasmin) while the remaining part of the protein does not carry copper (apoceruloplasmin). Ceruloplasmin acts as an acute phase reactant and may thus be increased by any inflammatory process; it may also rise in pregnancy and with the use of oestrogens and oral contraceptives. One also needs to remember that the normal range of serum ceruloplasmin is age-dependent: it is usually low in infants until the age of 6 months; in older children it may be somewhat higher than in adults. As explained previously, serum levels of ceruloplasmin are generally decreased in WD; however, this finding alone is unreliable because low serum ceruloplasmin may be seen without WD and serum ceruloplasmin may even be increased in severe WD and liver failure. Non-specific reductions of ceruloplasmin are usually associated with protein deficiency or any end-stage liver disease. Long-term parenteral nutrition may also lead to decreased levels of ceruloplasmin. Low serum ceruloplasmin is also a hallmark of Menkes’ disease, a very rare X-linked inborn error of metabolism that leads to a defect in copper transport due to mutations in ATP7A (Menkes 1999). Very rarely, one cannot measure serum ceruloplasmin at all. This aceruloplasminaemia is a very rare genetic disease caused by mutations in the ceruloplasmin gene; however, patients with aceruloplasminaemia develop iron and not copper overload (Harris 1998).
Most patients with WD have a serum ceruloplasmin lower than 20 μg/dL; this finding is diagnostic for WD however only when there are other findings such as a Kayser-Fleischer corneal ring. In one prospective screening study, ceruloplasmin was measured in 2867 patients presenting with liver disease: only 17 of them had reduced ceruloplasmin levels and only 1 of these subjects had WD (Cauza 1997). Thus decreased ceruloplasmin had a positive predictive value of only 6% in the 2867 patients tested. In two cohorts, about 20% of WD had normal ceruloplasmin and no Kayser-Fleischer rings (Steindl 1997, Gow 2000). Most reports, however, show that more than 90% of WD patients have a reduced serum ceruloplasmin (Walshe 1989, Lau 1990, Stremmel 1991). Measurement of ceruloplasmin as a single marker cannot reliably differentiate homozygotes from heterozygotes.
Serum copper
Corresponding to the decrease in serum ceruloplasmin, total serum copper is also usually decreased in WD. Similar to the diagnostic problems in interpreting ceruloplasmin data in WD patients with fulminant liver failure, serum copper may also be normal in this situation – even if serum ceruloplasmin is decreased. In acute liver failure, circulating copper may in fact be elevated because it is massively released from injured hepatocytes. If ceruloplasmin is reduced, a normal or elevated serum copper usually suggests that there is an increase in free serum copper (not bound to ceruloplasmin). The free copper concentration calculated from total copper and ceruloplasmin values has also been proposed as a diagnostic test and for monitoring of WD. It is elevated above 25 μg/dL in most untreated patients (normal values are below 10–15 μg/dL). The amount of copper associated with ceruloplasmin is 3.15 μg of copper per mg of ceruloplasmin. Thus free copper is the difference between the total serum copper in μg/dL and three times the ceruloplasmin concentration in mg/dL (Roberts 1998).
Increases in serum free copper, however, are not specific for WD and can be seen in all kinds of acute liver failure as well as in marked cholestasis (Gross 1985, Martins 1992). Thus, serum copper is not recommended as a primary tool for diagnosis of Wilson’s disease (Ferenci 2003, EASL 2012) (Table 2). Serum copper, however, is still recommended as a tool to monitor treatment (EASL 2012) (Table 3). The calculation of the free copper concentration critically depends on the adequacy of the methods used for measuring total serum copper and ceruloplasmin; often labs simply state that one of the tests is below a certain value, which makes it impossible to calculate the amount of free copper.
Urinary copper excretion
Most WD patients have an increase in urinary copper excretion above 100 μg/24 hours, which is thought to represent the increase in circulating free copper (not bound to ceruloplasmin). Some studies suggest that about 20% of WD patients may have 24-hr urinary copper excretion between 40–100 μg/24 h (Steindl 1997, Giacchino1997, Gow 2000, Roberts 2003). However, some increase in urinary copper excretion can be found in severe cholestasis, chronic active hepatitis and autoimmune hepatitis (Frommer 1981). It has been suggested that urinary copper excretion stimulated by penicillamine may be more useful than the non-stimulating test. In children, 500 mg of oral penicillamine is usually given at the beginning and then at 12 hours during the 24-hour urine collection. All WD children looked at had levels above 1600 μg copper/24 h and all patients with other liver diseases, including autoimmune hepatitis and cholestatic liver disease, had lower values. It is not clear whether this test has a similar discriminative power in adults where it has been used in various modifications (Tu 1967, Frommer 1981).
Hepatic copper concentration
Hepatic copper content above 250 μg/g dry weight liver is still the gold standard for diagnosis of WD and is not seen in heterozygotes or other liver diseases with the exception of Indian childhood cirrhosis (Martins 1992). Biopsies (larger than 1 cm in length) for measurements of hepatic copper determination should be taken with a disposable Tru-Cut needle, placed dry in a copper-free container and shipped frozen (Song 2000, Roberts 2003).
Radiolabelled copper
In WD, incorporation of radiolabelled copper into ceruloplasmin is significantly reduced. This test is rarely used because of the difficulty in obtaining the isotope and because of legal restrictions.
Liver biopsy findings
Histological findings in WD range from some steatosis and hepatocellular necrosis to the picture as seen in severe autoimmune hepatitis with fibrosis and cirrhosis. Patients diagnosed at a young age usually have extensive liver disease; older patients who first present with neurological symptoms often have abnormalities in liver biopsy as well (Stremmel 1991, Steindl 1997, Merle 2007). Detection of copper in hepatocytes, e.g. by staining with rhodamine using routine histochemistry, does not allow a diagnosis of WD (Geller 2000) (Figure 4).
Neurology and MRI of the CNS
Neurologic symptoms in WD include Parkinson’s-like abnormalities with rigidity, tremor and dysarthria. In more severely affected patients there may be muscle spasms, contractures, dysphonia, and dysphagia. In patients with pronounced neurological symptoms, magnetic resonance imaging (MRI) often identifies abnormalities in basal ganglia such as hyperintensity on T2-weighted imaging (Aisen 1995, van Wassanaer 1996). MRI of the CNS is superior to computed tomography to diagnose WD.
 Figure 4. Liver histology (rhodamine staining for copper) in a WD patient
Figure 4. Liver histology (rhodamine staining for copper) in a WD patient
Genetic Studies
The use of mutation analysis in WD is limited by the fact that more than 200 ATP7B mutations have been described (see www.medgen.med.ualberta.ca/database.html). When the mutation is known in a specific patient, gene analysis may be useful for family screening or prenatal analysis (Thomas 1995, Shab 1997, Loudianos 1994). Some populations in Eastern Europe show predominance of the H1069Q mutation (for literature see Roberts 2003). Recently genetic analysis is recommended as a basic tool for diagnosis of Wilson’s disease (Ferenci 2003, EASL 2012) (Table 2).
Table 2. EASL recommendations for diagnosis of Wilson’s disease (EASL 2012)| Wilson’s disease should be considered in any individual with liver abnormalities or neurological movement disorders of uncertain cause. Age alone should not be the basis for eliminating a diagnosis of Wilson’s disease. |
| Kayser-Fleischer rings should be sought by slit-lamp examination by a skilled examiner. The absence of Kayser-Fleischer rings does not exclude the diagnosis of Wilson’s disease, even in patients with predominantly neurological disease. |
| Neurologic evaluation and imaging of the brain, preferably by MR imaging, should be considered prior to treatment in all patients with neurologic Wilson’s disease and should be part of the evaluation of all patients presenting with neurological symptoms consistent with Wilson’s disease. |
| A low serum ceruloplasmin level should be taken as evidence for diagnosis of Wilson’s disease. Borderline levels require further evaluation. Serum ceruloplasmin within the normal range does not exclude the diagnosis. |
| Basal 24-hour urinary excretion of copper >1.6 μmol is typical in symptomatic patients. In children with mild hepatic disease basal 24-hour urinary excretion of copper can only be mildly elevated or may even be normal. |
| Hepatic parenchymal copper content >4 μmol/g dry weight provides critical diagnostic information and should be obtained in cases where the diagnosis is not straightforward and in younger patients. In untreated patients, normal hepatic copper content almost excludes Wilson’s disease. |
| Whole-gene sequencing is currently possible and haplotype analysis should be the primary mode for Wilson’s disease. |
Treatment
Before 1948, all patients with Wilson’s Disease died shortly after diagnosis. In 1948, intramuscular administration of the copper chelator BAL (dimercaprol) was introduced as the first treatment of WD (Cumming 1951, Denny-Brown 1951) followed by the oral chelators penicillamine (1955), trientine (1969) and tetrathiomolybdate (1984). Other treatment modalities include oral zinc salts (1961) and liver transplantation (1982). Today, most patients with WD remain on a lifelong pharmacologic therapy usually including a copper chelator and/or a zinc salt (Figure 5). LTX is reserved for fulminant liver failure and irreversible decompensation of liver cirrhosis. Patients with a successful LTX do not need WD treatment because LTX heals the biochemical defect. Today, most physicians use oral chelators for initial treatment of symptomatic patients; many physicians start therapy with penicillamine while some prefer trientine. Both drugs are probably equally effective, with trientine having fewer side effects. In patients with advanced neurological disease some authors recommend tetrathiomolybdate for primary therapy. Combination therapy of chelators and zinc salts might have additive effects, acting on both urinary copper excretion and its intestinal absorption. After removal of most accumulated copper and regression of the most severe clinical problems the chelator dose may be reduced and later replaced by zinc. Patients presenting without symptoms may be treated with a low dose of a chelator or with a zinc salt from the beginning. Compliance problems have been shown to regularly cause recurrence of symptomatic WD and may lead to fulminant liver failure, need for LTX or death.
Recent EASL guidelines summarise the treatment recommendations for Wilson’s disease (EASL 2012) (Table 3).
Table 3. Excerpts from the EASL recommendations for therapy of Wilson’s disease (EASL 2012)| Initial treatment for symptomatic patients with Wilson’s disease should include a chelating agent (D-penicillamine or trientine). Trientine may be better tolerated. Zinc may have a role in neurologically symptomatic patients. If zinc is used, careful monitoring of transaminases is needed, with changing to chelators if transaminases are increasing. |
| Treatment of presymptomatic patients or those with neurological disease on maintenance therapy can be accomplished with a chelating agent or with zinc. |
| Treatment is lifelong and should not be discontinued, unless liver transplantation is performed. |
| Patients should avoid foods and water with high concentrations of copper. |
| Patients with acute liver failure should be treated with liver transplantation when the revised King’s score is 11 or higher. |
| Patients with decompensated cirrhosis, unresponsive to chelation treatment, should be evaluated promptly for liver transplantation. |
| Treatment for Wilson’s disease should be continued during pregnancy, but dose reduction is advisable for D-penicillamine and trientine. |
| For routine monitoring, serum copper and ceruloplasmin, liver enzymes and function test, blood count and urine analysis as well as physical and neurological examinations should be performed at least twice annually. |
| The 24-hour urinary copper excretion on medication and after 2 days of cessation of therapy should be measured at least yearly. The estimated serum non-ceruloplasminbound copper may be a useful monitoring parameter. |
Penicillamine. Penicillamine was the first oral copper chelator shown to be effective in WD (Walshe 1955). Total bioavailability of oral penicillamine ranges between 40 and 70% (Bergstrom 1981). Many studies have shown that penicillamine reduces copper accumulation and provides clinical benefit in WD (Walshe 1973, Grand 1975, Sternlieb 1980). Signs of liver disease often regress during the initial 6 months of treatment. Non-compliance has been shown to cause progression of liver disease, liver failure, death and LTX (Scheinberg 1987). However, neurological symptoms may deteriorate at the start of penicillamine treatment; it remains controversial how often this neurological deterioration occurs and whether it is reversible; the rate of neurological worsening ranges from 10–50% in different cohorts (Brewer 1987, Walshe 1993). Some authors even recommend not using penicillamine in WD patients with neurological disease (Brewer 2006). Penicillamine is associated with many side effects that lead to its discontinuation in up to 30% of patients (for literature see Roberts 2003). An early sensitivity reaction may occur during the first three weeks including fever, cutaneous exanthema, lymphadenopathy, neutropenia, thrombocytopenia and proteinuria. In such early sensitivity, penicillamine should be replaced by trientine immediately. Nephrotoxicity is another frequent side effect of penicillamine, which occurs later and includes proteinuria and signs of tubular damage. In this case penicillamine should be immediately discontinued. Penicillamine may also cause a lupus-like syndrome with hematuria, proteinuria, positive antinuclear antibody and Goodpasture’s Syndrome. More rarely the drug can damage the bone marrow leading to thrombocytopenia or total aplasia. Dermatologic side effects include elastosis perforans serpiginosa, pemphigoid lesions, lichen planus and aphthous stomatitis. There have also been reports of myasthenia gravis, polymyositis, loss of taste, reduction of IgA and serous retinitis due to administration of penicillamine.
In order to minimise its side effects pencillamine should be started at 250 mg daily; the dose may be increased in 250 mg steps every week to a maximal daily amount of 1000 to 1500 mg given in 2 to 4 divided doses daily (Roberts 2003). Maintenance doses range from 750 to 1000 mg/d given as 2 divided doses. In children the dose is 20 mg/kg/d given in 2 or 3 divided doses. Penicillamine should be given 1 hour before or 2 hours after meals because food may inhibit its absorption. After starting penicillamine therapy serum ceruloplasmin at first may decrease. Treatment success is checked by measuring 24-hr urinary copper that should range between 200–500 μg/day. In the long run, ceruloplasmin should increase and free copper should regress towards normal with penicillamine therapy (Roberts 2003).
Trientine (triene). The chemical structure of the copper chelator trientine (triethylene tetramine dihydrochloride, AKA triene) differs from penicillamine. Trientine has usually been used as an alternative or substitute for penicillamine, in particular when penicillamine’s major side effects are not tolerable (Walshe 1982). Triene only rarely has side effects. Similar to penicillamine, long-term treatment with trientine may cause hepatic iron accumulation in persons with WD. Trientine is poorly absorbed from the gastrointestinal tract, and only 1% appears in the urine (Walshe 1982). Doses range from 750 to 1500 mg/d given in 2 or 3 divided doses; 750 or 1000 mg are given for maintenance therapy (Roberts 2003). In children, a dose of 20 mg/kg/d is recommended. Similar to penicillamine, trientine should be given 1 hour before or 2 hours after meals. The effectiveness of copper chelation by triene is measured as described for penicillamine. Triene chelates several metals such as copper, zinc and iron by urinary excretion and it effectively removes accumulated copper from various organs in persons with WD as well as in severe liver disease (Walshe 1979, Scheinberg 1987, Santos 1996, Saito 1991). It is still unclear whether penicillamine is a more effective copper chelator when compared to triene; probably the difference in effectiveness is small (Walshe 1973, Sarkar 1977). Potential deterioration of neurological disease may also be seen after starting triene therapy; the worsening however is less frequent and less pronounced than that seen after starting with penicillamine.
Zinc. Most physicians substitute penicillamine or triene with zinc for maintenance therapy when most copper accumulation has been removed. Zinc can also be given as initial therapy in asymptomatic patients diagnosed by family screening. A recent report however shows that WD symptoms may occur despite zinc prophylaxis in asymptomatic patients (Mishra 2008). In a recent study from India, 45 WD patients were on both penicillamine and zinc sulfate. The majority of patients (84%) had neuropsychiatric manifestations. The mean duration of treatment with penicillamine and zinc, before stopping penicillamine, was 107 months. All patients had to stop penicillamine due to the financial burden. The patients then only received zinc sulfate for 27 months and 44 of the 45 patients (98%) remained stable. Only one patient reported worsening in dysarthria (Sinha 2008). Zinc does not act as an iron chelator but inhibits intestinal copper absorption and has also been suggested to bind free toxic copper (Brewer 1983, Schilksky 1989, Hill 1987). Zinc rarely has any side effects. It is still unclear whether zinc as monotherapy is an effective “decoppering” agent in symptomatic patients. There are some hints that hepatic copper may accumulate despite zinc therapy including reports about hepatic deterioration with a fatal outcome (Lang 1993, Walshe 1995). Therefore some authors use zinc in combination with a chelator. Neurological deterioration is rather rare under zinc therapy (Brewer 1987, Czlonkowska 1996). The recommended doses of zinc vary in the literature: according to AASLD practice guidelines dosing is in milligrams of elemental zinc (Roberts 2003). For larger children and adults, 150 mg/d is administered in divided doses. Compliance with doses given thrice daily may be problematic; zinc has to be taken at least twice daily to be effective (Brewer 1998). Other authors recommend using zinc sulfate at 150 mg thrice daily as a loading dose and 100 mg thrice daily for maintenance. Further recommendations suggest giving 50 mg as zinc acetate thrice daily in adults. The type of zinc salt used has been thought to make no difference with respect to efficacy (Roberts 2003). However, zinc acetate has been suggested to cause the least gastrointestinal discomfort. When zinc is combined with a chelator the substances should be given at widely spaced intervals, potentially causing compliance problems. Effectiveness of the zinc treatment should be checked as described for penicillamine and zinc (Roberts 2003).
Tetrathiomolybdate. Tetrathiomolybdate is an experimental copper chelator not approved by FDA or EMA. It has been suggested as the initial treatment of WD patients with neurological involvement. Early reports say that tetrathiomolybdate stabilises the neurological disease and reduces circulating free copper in a matter of weeks (Brewer 1994, Brewer 1996). A more recent randomised study supports this view and also suggests that zinc monotherapy is insufficient for treatment of neurological WD (Brewer 2006).
Vitamin E, other antioxidants and diet. Since serum and hepatic concentrations of vitamin E levels may be reduced in WD (von Herbay 1994, Sokol 1994) it has been suggested to complement vitamin E intake. Some authors have also recommended taking other antioxidants; studies have not proven their effectiveness as yet.
WD patients should avoid food with high copper content (nuts, chocolate, shellfish, mushrooms, organ meats, etc). Patients living in older buildings should also check whether the water runs through copper pipes. Such dietary and lifestyle restrictions do not replace chelator or zinc therapy (Roberts 2003).
Fulminant hepatic failure and LTX. Most WD patients with fulminant liver failure need LTX urgently in order to survive (Sokol 1985, Roberts 2003). However, in a long-term cohort study only two patients died prior to LTX being available (Stremmel 1991). It is a difficult clinical question whether WD patients with liver failure can survive without LTX. The prognostic score used to help with this difficult decision includes bilirubin, AST, and INR (Nazer 1986). In any case, WD patients with signs of fulminant liver failure need to be transferred immediately (same day!) to a transplant centre.
WD patients with a chronic course of decompensated cirrhosis follow the usual rules for LTX. LTX cures the metabolic defects and thus copper metabolism returns to normal afterwards (Groth 1973). Prognosis for WD after LTX is excellent, in particular when patients survive the first year (Eghtesad 1999). It is still unclear under which circumstances LTX may be helpful for WD patients with neurological complications, which do not respond to drug therapy. In some patients CNS symptoms regress after LTX while other patients do not improve (for literature see Brewer 2000).
Asymptomatic Patients. All asymptomatic WD subjects – usually identified by family screening – need to be treated by chelators or zinc in order to prevent life-threatening complications (Walshe 1988, Brewer 1989, Roberts 2003). It is unclear whether therapy should begin in children under the age of 3 years.
Maintenance Therapy. After initial removal of excessive copper by chelators, some centres replace the chelators with zinc for maintenance therapy. It is unclear when such change is advisable and whether it might be better to reduce the dose of chelators instead of replacing them with zinc. It is generally accepted that replacement of chelators with zinc should only be done in patients who are clinically stable for some years, have normal aminotransferase and liver function, a normal free copper concentration and a 24-hr urinary copper repeatedly in the range of 200–500 μg while on chelators (Roberts 2003). Long-term treatment with zinc may be associated with fewer side effects than chelator treatment. Many patients on trientine, however, do have significant side effects, and this author believes one does need to replace trientine with zinc in such patients. In any case, therapy either with a chelator or with zinc needs to be maintained indefinitely; any interruption may lead to lethal liver failure (Walshe 1986, Scheinberg 1987).
Pregnancy. Treatment must be maintained during pregnancy because an interruption has been shown to carry a high risk of fulminant liver failure (Shimono 1991). Maintenance therapy with chelators (penicillamine, trientine) or with zinc usually results in a good outcome for mother and child, although birth defects have (rarely) been documented (for literature see Sternlieb 2000). It is recommended that the doses of both chelators be reduced, if possible by about 50%, in particular during the last trimester to avoid potential problems in wound healing (Roberts 2003). Zinc does not need to be reduced.
Monitoring of treatment
Monitoring should be done closely during initial treatment in all WD patients to look for efficacy (Table 4) and side effects. During the maintenance phase patients should be checked at least twice a year.
Table 4. Monitoring the treatment efficacy in WD| Clinical improvement (neurologic features, liver disease, hematology) |
| Regression of Kayser-Fleischer Ring |
| Circulating free copper <10 µl/dL |
| 24-hr urinary copper excretion (200–500 µg/day on chelators) |
| Decrease in liver copper content |
Clinical examinations include neurological, ophthalmologic and psychiatric consultations (Figure 5). Patients with liver involvement need to be checked carefully for signs of liver failure.
Laboratory tests include measurements of serum copper and ceruloplasmin, calculation of free (non-ceruloplasmin-bound) copper (see above), and 24-hr urinary copper excretion (Roberts 2003). While on chelating therapy 24-hr urinary copper excretion should initially range between 200 and 500 μg; such a value can also suggest that the patient is adherent to the drug. After removal of copper accumulation, urinary copper excretion may be lower. Prognosis of WD is dependent on the initial severity of the disease and then on adherence to the life-long treatment. Patients treated prior to severe and potentially irreversible neurological and hepatic complications have a good prognosis approaching a normal life expectancy (Figure 6). Irreversible liver disease often can be treated successfully by LTX while some patients with severe neurological disease do not get better despite optimal therapy.
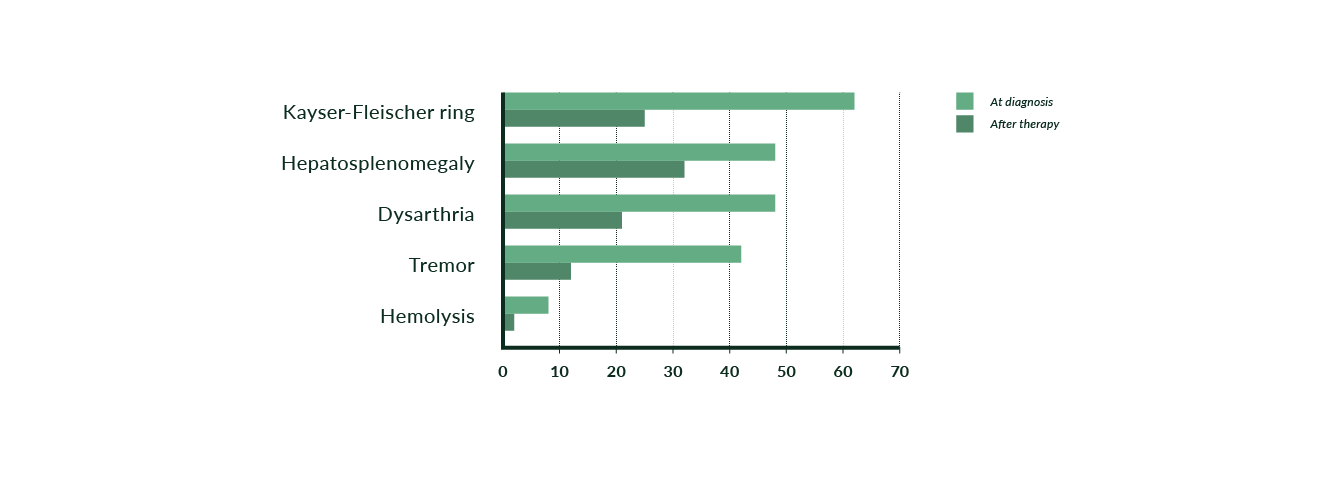 Figure 5. Findings prior to and after beginning chelating therapy in 53 WD patients (modified from Stremmel 1991)
Figure 5. Findings prior to and after beginning chelating therapy in 53 WD patients (modified from Stremmel 1991)
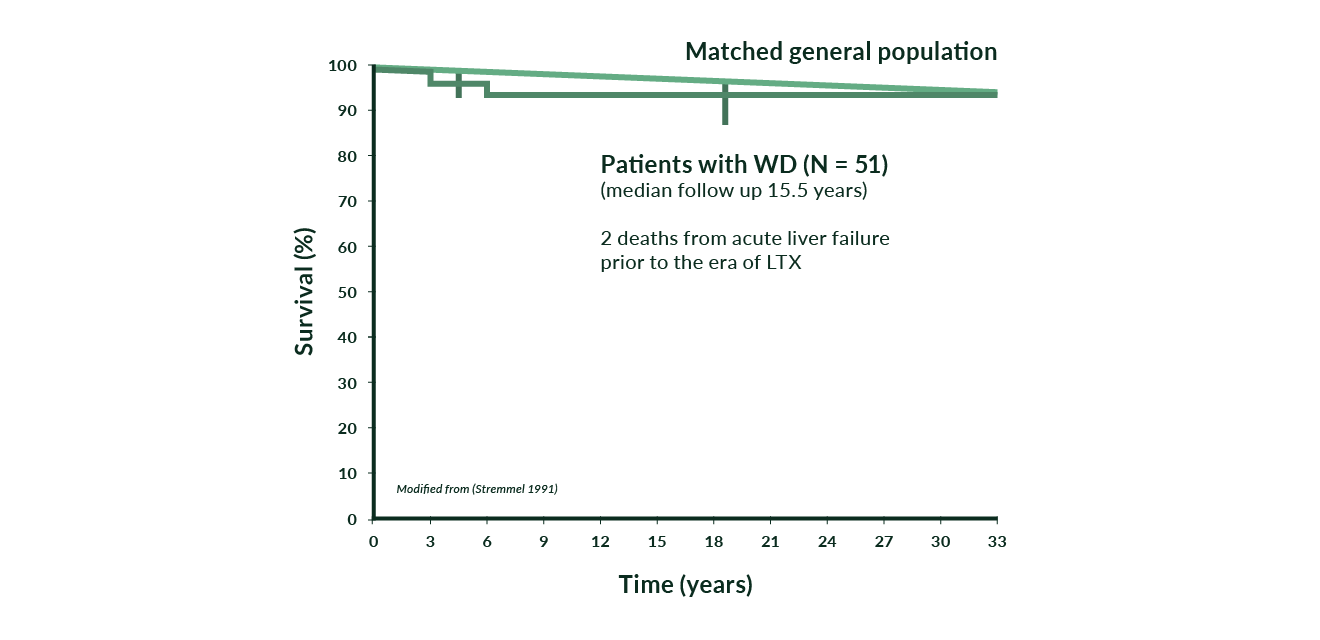 Figure 6. Cumulative survival in 51 WD patients versus a matched general population (modified from Stremmel 1991)
Figure 6. Cumulative survival in 51 WD patients versus a matched general population (modified from Stremmel 1991)
References
Aisen AM, Martel W, Gabrielsen TO, et al. Wilson disease of the brain: MR imaging. Radiology 1985;157:137-41.
Azizi E, Eshel G, Aladjem M. Hypercalciuria and nephrolithiasis as a presenting sign in Wilson disease. Eur J Pediatr 1989;148:548-9.
Bergstrom RF, Kay DR, Harkcom TM, Wagner JG. Penicillamine kinetics in normal subjects. Clin Pharmacol Ther 1981;30:404-13.
Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991;100:1129-34.
Brewer GJ, Hill GM, Prasad AS, Cossack ZT, Rabbani P. Oral zinc therapy for Wilson’s disease. Ann Intern Med 1983;99:314-9.
Brewer GJ, Terry CA, Aisen AM, Hill GM. Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch Neurol 1987;44:490-3.
Brewer GJ, Yuzbasiyan-Gurkan V, Young AB. Treatment of Wilson’s disease. Semin Neurol 1987;7:209-20.
Brewer GJ, Yuzbasiyan-Gurkan V, Lee DY, Appelman H. Treatment of Wilson’s disease with zinc. VI. Initial treatment studies. J Lab Clin Med 1989;114:633-8.
Brewer GJ, Dick RD, Johnson V, et al. Treatment of Wilson’s disease with ammonium tetrathiomolybdate. I. Initial therapy in 17 neurologically affected patients. Arch Neurol 1994;51:545-54.
Brewer GJ, Johnson V, Dick RD, Kluin KJ, Fink JK, Brunberg JA. Treatment of Wilson disease with ammonium tetrathiomolybdate. II. Initial therapy in 33 neurologically affected patients and follow-up with zinc therapy. Arch Neurol 1996;53:1017-25.
Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J Lab Clin Med 1998;132:264-78.
Brewer GJ, Askari F. Transplant livers in Wilson’s disease for hepatic, not neurologic, indications. Liver Transpl 2000;6:662-4.
Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate. IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol 2006;63:521-7 .
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 1993;5:327-37.
Cairns JE, Williams HP, Walshe JM. “Sunflower cataract” in Wilson’s disease. Br Med J 1969;3:95-6.
Cauza E, Maier-Dobersberger T, Polli C, Kaserer K, Kramer L, Ferenci P. Screening for Wilson’s disease in patients with liver diseases by serum ceruloplasmin. J Hepatol 1997;27:358-62.
Chu CC, Huang CC, Chu NS. Recurrent hypokalemic muscle weakness as an initial manifestation of Wilson’s disease. Nephron 1996;73:477-9.
Cumings JN. The effect of BAL in hepatolenticular degeneration. Brain 1951;74:10-22.
Czlonkowska A, Gajda J, Rodo M. Effects of long-term treatment in Wilson’s disease with D-penicillamine and zinc sulphate. J Neurol 1996; 243:269-73.
Denny-Brown D, Porter H. The effect of BAL (2,3 dimercaptopropanol) on hepatolenticular degeneration (Wilson’s disease). NEngl J Med 1951;245:917-25.
EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol 2012;56: 671–685.
Eghtesad B, Nezakatgoo N, Geraci LC, et al. Liver transplantation for Wilson’s disease: a single-center experience. Liver Transpl Surg 1999;5:467-74.
Emre S, Atillasoy EO, Ozdemir S, et al. Orthotopic liver transplantation for Wilson’s disease: a single-center experience. Transplantation 2001;72:1232-6.
Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003;23:139–142.
Ferenci P, Czlonkowska A, Merle U, et al. Late-onset Wilson’s disease. Gastroenterology 2007;132:1294-8.
Ferlan-Marolt V, Stepec S. Fulminant Wilsonian hepatitis unmasked by disease progression: report of a case and review of the literature. Dig Dis Sci 1999;44:1054-8.
Fleischer B. Ueber einer der “Pseudosklerose” nahestehende bisher unbekannte Krankheit (gekennzeichnet durch Tremor, psychische Stoerungen, braeunliche Pigmentierung bestimmter Gewebe, insbesondere der Hornhautperipherie, Lebercirrhose). Deutsch Z Nerven Heilk 1912;44:179-201.
Frommer DJ. Urinary copper excretion and hepatic copper concentrations in liver disease. Digestion 1981;21:169-78.
Frydman M. Genetic aspects of Wilson’s disease. J Gastroenterol Hepatol 1990;5:483-90.
Geller SA, Petrovic LM, Batts KB, et al. Histopathology of end-stage Wilson disease. Mod Pathol 2000; 13:184A.
Giacchino R, Marazzi MG, Barabino A, et al. Syndromic variability of Wilson’s disease in children. Clinical study of 44 cases. Ital J Gastroenterol Hepatol 1997;29:155-61.
Grand RJ, Vawter GF. Juvenile Wilson disease: histologic and functional studies during penicillamine therapy. J Pediatr 1975;87:1161-70.
Gross JB, Jr., Ludwig J, Wiesner RH, McCall JT, LaRusso NF. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology 1985;89:272-8.
Groth CG, Dubois RS, Corman J, et al. Metabolic effects of hepatic replacement in Wilson’s disease. Transplant Proc 1973;5:829-33.
Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. Diagnosis of Wilson’s disease: an experience over three decades. Gut 2000;46:415-9.
Harris ZL, Klomp LW, Gitlin JD. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr 1998;67:972S-977S.
Hill GM, Brewer GJ, Prasad AS, Hydrick CR, Hartmann DE. Treatment of Wilson’s disease with zinc. I. Oral zinc therapy regimens. Hepatology 1987;7:522-8.
Kalach N, Seidman EG, Morin C, et al. Acute liver failure from Wilson’s disease in a five year-old child. Can J Gastroenterol 1993;7:610-2.
Lang CJ, Rabas-Kolominsky P, Engelhardt A, Kobras G, Konig HJ. Fatal deterioration of Wilson’s disease after institution of oral zinc therapy. Arch Neurol 1993;50:1007-8.
Lau JY, Lai CL, Wu PC, Pan HY, Lin HJ, Todd D. Wilson’s disease: 35 years’ experience. Q J Med 1990;75:597-605.
Loudianos G, Figus AL, Loi A, et al. Improvement of prenatal diagnosis of Wilson disease using microsatellite markers. Prenat Diagn 1994;14:999-1002.
Martins da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, Mieli-Vergani G. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology 1992;15: 609-15.
Menkes JH. Menkes disease and Wilson disease: two sides of the same copper coin. Part I: Menkes disease. Europ J Paediatr Neurol 1999;3: 147-58.
Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut 2007;56:115-20.
Milkiewicz P, Saksena S, Hubscher SG, Elias E. Wilson’s disease with superimposed autoimmune features: report of two cases and review. J Gastroenterol Hepatol 2000;15:570-4.
Mishra D, Kalra V, Seth R. Failure of prophylactic zinc in Wilson disease. Indian Pediatr 2008;45:151-3.
Nakada SY, Brown MR, Rabinowitz R. Wilson’s disease presenting as symptomatic urolithiasis: a case report and review of the literature. J Urol 1994;152:978-9.
Nazer H, Ede RJ, Mowat AP, Williams R. Wilson’s disease: clinical presentation and use of prognostic index. Gut 1986;27:1377-81.
Petrukhin K, Fischer SG, Pirastu M, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet 1993;5:338-43.
Rector WG, Uchida T, Kanel GC, Redeker AG, Reynolds TB. Fulminant hepatic and renal failure complicating Wilson’s disease. Liver 1984;4:341-7.
Roberts EA, Cox DW. Wilson disease. Baillieres Clin Gastroenterol 1998;12:237-56.
Roberts EA, Schilsky ML. A Practice Guideline on Wilson Disease. Hepatology 2003;37:1475-92.
Saito T. Presenting symptoms and natural history of Wilson disease. Eur J Pediatr 1987;146:261-5.
Saito H, Watanabe K, Sahara M, Mochizuki R, Edo K, Ohyama Y. Triethylene-tetramine (trien) therapy for Wilson’s disease. Tohoku J Exp Med 1991;164:29-35.
Sallie R, Katsiyiannakis L, Baldwin D, et al. Failure of simple biochemical indexes to reliably differentiate fulminant Wilson’s disease from other causes of fulminant liver failure. Hepatology 1992;16:1206-11.
Santos Silva EE, Sarles J, Buts JP, Sokal EM. Successful medical treatment of severely decompensated Wilson disease. J Pediatr 1996;128:285-7.
Sarkar B, Sass-Kortsak A, Clarke R, Laurie SH, Wei P. A comparative study of in vitro and in vivo interaction of D-penicillamine and triethylene-tetramine with copper. Proc R Soc Med 1977;70:13-8.
Scheinberg IH, Gitlin D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease). Science 1952;116:484-5.
Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med 1987;317:209-13.
Schilsky M, Blank RR, Czaja MJ, Scheinberg IH, Stockert RJ, Sternlieb I. Hepatocellular copper toxicity and its attenuation by zinc. J Clin Invest 1989;84:1562-8.
Schilsky ML, Scheinberg IH, Sternlieb I. Prognosis of Wilsonian chronic active hepatitis. Gastroenterology 1991;100:762-7.
Schilsky ML, Scheinberg IH, Sternlieb I. Liver transplantation for Wilson’s disease: indications and outcome. Hepatology 1994;19:583-7.
Scott J, Gollan JL, Samourian S, Sherlock S. Wilson’s disease, presenting as chronic active hepatitis. Gastroenterology 1978;74:645-51.
Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997;61:317-28.
Shimono N, Ishibashi H, Ikematsu H, et al. Fulminant hepatic failure during perinatal period in a pregnant woman with Wilson’s disease. Gastroenterol Jpn 1991;26:69-73.
Sinha S, Taly AB. Withdrawal of penicillamine from zinc sulphate-penicillamine maintenance therapy in Wilson’s disease: promising, safe and cheap. J Neurol Sci 2008;264:129-32.
Sokol RJ, Francis PD, Gold SH, Ford DM, Lum GM, Ambruso DR. Orthotopic liver transplantation for acute fulminant Wilson disease. J Pediatr 1985;107:549-52.
Sokol RJ, Twedt D, McKim JM, et al. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology 1994;107:1788-98.
Song YM, Chen MD. A single determination of liver copper concentration may misdiagnose Wilson’s disease. Clin Biochem 2000;33:589-90.
Steindl P, Ferenci P, Dienes HP, et al.Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212-218.
Sternlieb I. Copper and the liver. Gastroenterology 1980;78:1615-28.
Sternlieb I. Perspectives on Wilson’s disease. Hepatology 1990;12:1234-9.
Sternlieb I. Wilson’s disease and pregnancy. Hepatology 2000;31:531-2.
Stremmel W, Meyerrose KW, Niederau C, Hefter H, Kreuzpaintner G, Strohmeyer G. Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med 1991;115:720-6.
Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homologyto the Menkes disease gene. Nat Genet 1993;5:344-50.
Tauber J, Steinert RF. Pseudo-Kayser-Fleischer ring of the cornea associated with non-Wilsonian liver disease. A case report and literature review.Cornea 1993;12:74-7.
Thomas GR, Roberts EA, Walshe JM, Cox DW. Haplotypes and mutations in Wilson disease. Am J Hum Genet 1995;56:1315-9.
Tu JB, Blackwell RQ. Studies on levels of penicillamine-induced cupriuresis in heterozygotes of Wilson’s disease. Metabolism 1967;16:507-13.
van Wassenaer-van Hall HN, van den Heuvel AG, Algra A, Hoogenraad TU, Mali WP. Wilson disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology 1996;198:531-6.
von Herbay A, de Groot H, Hegi U, Stremmel W, Strohmeyer G, Sies H. Low vitamine E content in plasma of patients with alcoholic liver disease, haemochromatosis and Wilson’s disease. J Hepatol 1994;20:41-6.
Walshe JM. Wilson’s disease. New oral therapy. Lancet 1956;i:25-6.
Walshe JM. Copper chelation in patients with Wilson’s disease. A comparison of penicillamine and triethylene tetramine dihydrochloride. Q J Med 1973;42:441-52.
Walshe JM. The management of Wilson’s disease with trienthylene tetramine 2HC1 (Trien 2HC1). Prog Clin Biol Res 1979;34:271-80.
Walshe JM. Treatment of Wilson’s disease with trientine (triethylenetetramine) dihydrochloride. Lancet 1982;1:643-7.
Walshe JM, Dixon AK. Dangers of non-compliance in Wilson’s disease. Lancet 1986;1:845-7.
Walshe JM. Diagnosis and treatment of presymptomatic Wilson’s disease. Lancet 1988;2:435-7.
Walshe JM. Wilson’s disease presenting with features of hepatic dysfunction: a clinical analysis of eighty-seven patients. Q J Med 1989;70:253-63.
Walshe JM, Yealland M. Chelation treatment of neurological Wilson’s disease. Q J Med 1993;86:197-204.
Walshe JM, Munro NA. Zinc-induced deterioration in Wilson’s disease aborted by treatment with penicillamine, dimercaprol, and a novel zero copper diet. Arch Neurol 1995;52:10-1.
Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912;34:295-507.
Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr 2000;137:719-22.
Yamaguchi Y, Heiny ME, Gitlin JD. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun 1993;197:271-7.