
22. NAFLD and NASH
Claus Niederau
Introduction
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are the most common chronic liver diseases in the West (Tayama 2012, Cusi 2012). They are closely associated with obesity, type 2 diabetes mellitus and metabolic syndrome. The epidemics of diabetes and obesity have also fueled an increasing prevalence of fatty liver disease (Tayama 2012, Cusi 2012). Both NAFLD and NASH are associated with an often asymptomatic elevation of serum ALT and gamma GT. Ultrasound monitoring can suggest the presence of a fatty infiltration of the liver; differentiation between NAFLD and NASH, however, often requires a liver biopsy. Such differentiation is important because NASH is associated with a much higher risk of liver fibrosis and cirrhosis than NAFLD. However, publications also show that most patients with fatty liver disease die from cardiovascular disease and not from liver disease (Pisto 2014, Treeprasertsuk 2013, Haflidadottir 2014, Kim 2013). NAFLD-related hepatocellular carcinoma (HCC) is a relevant complication and may occur also in the absence of cirrhosis (Piscaglia 2015), but is still less prevalent than HCV-related HCC (Beste 2015, Piscaglia 2015, Mittal 2015a, Mittal 2015b, White 2012). Globally, only the HCV-related HCC has increased in the last 15 years, but not NAFL-related HCC (Naghavi 2015). Although NAFLD is a growing cause for liver transplantation (LTX), HCV is still the leading cause for LTX (Wong 2014).
Prevalence
NAFLD is present in 20 to 40% of the general population in industrialised countries and is the most prevalent chronic liver disease (Browning 2004, Chitturi 2004, McCullough 2005). It is more prevalent in obese and diabetic subjects (Bellentani 1994, Wanless 1990, Clark 2002, Chitturi 2004). Among all subjects with NAFLD, features of non-alcoholic steatohepatitis (NASH) can be seen in 10-20%. The prevalence of NASH in Western countries is approximately 2-6%. In the US, NASH is estimated to affect 5-6% of the general population (McCullough 2005). It has been suggested that NASH accounts for more than 50% of cryptogenic cirrhosis (Ratziu 2002). NAFLD may progress to NASH with fibrosis, cirrhosis, and hepatocellular carcinoma (Marchesini 2003, Caldwell 2004). The term NASH was introduced in a description of 20 Mayo Clinic patients with a hitherto unnamed disease associated with hepatomegaly, abnormal ALT, a fatty liver histology, lobular hepatitis, and fibrosis mimicking alcoholic hepatitis in the absence of alcohol intake (Ludwig 1980); most patients had obesity and diabetes mellitus.
Demographics and risk factors
In the US, NAFLD is 3-5 times more prevalent in men than in women; such differences in gender might partly be explained by the fact that men have a higher BMI and that some male patients with NAFLD drink more alcohol than they report (Schwimmer 2005, Bahcecioglu 2006, Loguercio 2001). The NAFLD prevalence in the US is particularly high in people of Hispanic (28%) or Asian (20-30%) origin (Schwimmer 2005, Weston 2005). Due to the dramatic increase in obesity in the US and other industrialised countries, there is also a dramatic increase in the prevalence of NAFLD and NASH (Tayama 2012, Cusi 2012). In the US almost 50% of obese boys have NAFLD (Schwimmer 2005). In many countries more than 80% of NAFLD patients have an increased BMI and 30-40% are obese; approximately 50% show signs of insulin resistance, 20-30% have type 2 diabetes, 80% show hyperlipidaemia, and 30-60% have arterial hypertension. Correspondingly there is a strong association between NAFLD and NASH and the metabolic syndrome throughout the world (Marchesini 1999, Bedogni 2005). In comparison with NAFLD patients, NASH patients are older, more obese and more often have high serum liver enzymes, diabetes mellitus and metabolic syndrome (Ratziu 2002, Adams 2005, Hamaguchi 2005, Fassio 2004, Tayama 2012, Cusi 2012).
Pathogenesis
The degree of fatty infiltration in NAFLD is graded according to the percentage of hepatocytes with fat deposits: mild NAFLD involves less than 30% hepatocytes, moderate NAFLD up to 60%, and severe NAFLD above 60% (Ploeg 1993). NAFLD may regress if the cause is eliminated. NASH is associated with insulin resistance, increased circulating levels of leptin, adiponectin, tumour necrosis factor and some interleukins (Friedman 1998, Marra 2004). A meta-analysis confirmed that circulating leptin levels were higher in patients with NAFLD than in controls. Higher levels of circulating leptin were associated with increased severity of NAFLD, and the association remained significant after the exclusion of studies involving paediatric or adolescent populations and morbidly obese individuals being referred for bariatric surgery (Polyzos 2016). It is thought that there is an increased flow of free fatty acids from visceral fat to the liver contributing to abnormalities in intracellular lipid metabolism (Hashimoto 1999, Vendemiale 2001). Insulin resistance and increased free fatty acids may both affect mitochondrial oxidation of fatty acids causing free radical generation in hepatocytes (Grattagliano 2003). Thus, NASH is caused by two mechanisms or toxic “hits”: the first mechanism is the hepatic accumulation of triglycerides (NAFLD) due to insulin resistance and the second is thought to be the generation of free radicals with subsequent release of mediators and cytokines (McCullough 2006).
Insulin resistance has been closely linked to non-alcoholic fatty liver disease in both clinical trials and laboratory-based studies (McCullough 2006, Marchesini 2001, Sanyal 2001). The actual process by which NAFLD turns into NASH however remains ill-defined despite this double-hit theory. Likely, genetic factors (similar to those responsible for the metabolic syndrome) as well as exogenic factors (like drugs, moderate amounts of alcohol, and other toxins) may contribute to the evolution of NAFLD into NASH. The role of hepatic iron in the progression of NASH remains controversial, but in some patients, iron may have a role in the pathogenesis of NASH by promoting oxidative stress (Lee 1995, George 1998, Bonkowsky 1999, Younoussi 1999).
Human genetic factors
Until recently, the genetic determinants of the pathogenesis and disease progression of NAFLD remained obscure. In 2008, two genome-wide association (GWAS) studies linked the rs738409 polymorphism (IL48M) of patatin-like phospholipase domain containing 3 (PNPLA3) with hepatic fat content and ALT levels (Romeo 2008, Yuan 2008). Several later studies and a recent meta-analysis have corroborated such association between the IL48M polymorphism and NAFLD in almost all ethnic groups, and in adults, children and adolescents (Kontronen 2009, Sookoian 2009, Valenti 2010, Romeo 2010a, Romeo 2010b, Rotman 2010, Hotta 2010, Speliote 2010, Davis 2010, Santoro 2010a, Santoro 2010b, Sookoian 2011, Guidice 2011, Lin 2011, Valenti 2012, Peng 2012, Nobili 2013, Kitamoto 2013, Hernaez 2013, Shang 2014, DiStefano 2014, Baclig 2014). Further studies suggest that the 148M variant is an important risk factor for accumulation of hepatic steatosis in particular when additional factors are present such as free fatty acid release, insulin resistance, visceral obesity, increase in lipogenesis, and changes in lipid metabolism (Guidice 2011, He 2010, Davis 2010, Santoro 2010a, Santoro 2010b, Sevastianova 2012, Valenti 2012, Dongiovanni 2013).
IL48M polymorphism also predisposes to cirrhosis (Shen 2014) and hepatocellular carcinoma (Falleti 2011, Burza 2012, Trepo 2012, Valenti 2013, Nault 2014, Trepo 2014). Potential mechanisms involved have recently been reviewed and are not discussed in detail here (Dongiovanni 2013, Nault 2014, Shen 2014). All recent data suggest that the 148M PNPLA3 polymorphism favours hepatic carcinogenesis in steatohepatitis as well as in other liver diseases, and the mechanism is partly independent of the predisposition towards fibrogenesis and cirrhosis (Nault 2014, Shen 2014).
The association of the rs738409 SNP in the adiponutrin/PNPLA3 gene with alcoholic and non-alcoholic fatty liver disease and with intracellular triglyceride accumulation is seen also in European populations (Trepo 2014). In parallel, recent GWAS identified other loci, including neurocan (NCAN), associated with liver fat content and progression of non-alcoholic fatty liver disease (Speliotes 2011). Some of these variants were associated with distinct changes in serum lipid levels, which suggest different and specific impacts on lipid metabolism and NAFLD progression. In particular, the T allele of NCAN rs2228603 was suggested to induce hepatic triglyceride accumulation (Speliotes 2011). In a recent study (Nischalke 2014) the NCAN rs2228603 and the PNPLA3 IL48M variants were independently associated with increased prevalence of HCC in two genotyped cohorts and were used to stratify patients for risk of liver cancer. This data underlines the importance of steatosis in liver carcinogenesis (Trepo 2014). Other recent studies show similar findings (Sookoian 2014, Liu 2014, Di Stefano 2014).
The IL48M polymorphism is also a major risk factor for steatosis in chronic hepatitis C virus infection, in particular in non-genotype 3 infections (Cai 2011, Valenti 2011, Müller 2011, Trepo 2011, De Nicola 2014). These studies have further proven the association between the IL48M PNPLA3 variant and fibrosis progression in patients with chronic hepatitis C virus infection.
Recent data has also show evidence that the 148M variant predisposes to steatosis and thereby to progressive fibrosis in chronic hepatitis B, haemochromatosis, primary sclerosing cholangitis and alcoholic liver disease (Valenti 2012, Trepo 2012, Friedrich 2013, Vigano 2013).
Despite the recent finding that polymorphisms in the adiponutrin/PNPLA3 gene modify steatosis and fibrosis, large GWAS studies were much less successful in identifying human genetic factor leading to obesity, fatty liver, insulin resistance, and diabetes mellitus (Holzapfel 2010, Delahanty 2012). Lifestyle factors were correlated with BMI more closely than genetic factors (Holzapfel 2010).
Microbiome
The gut microbiota, now also called the gut microbiome, is involved in the pathophysiology of non-alcoholic fatty liver disease as well as in obesity and the metabolic syndrome. All the metabolic products generated by the intestinal microbiome first enter the liver. Studies with germ-free mice have shown that inoculation of microbiota from conventionally raised fat mice results in obesity and fatty liver (Bäckhed 2009). Genetically obese (ob/ob) mice have a decreased ratio of bacteroides versus firmicutes compared with lean (ob/+ and +/+ wild-type) mice (Ley 2009). Inoculation of gut microbiota from these obese mice (ob/ob) to germ-free mice led to an obese phenotype (Turnbaugh 2006). Similar effects occur when such mice are fed a Western diet or are inoculated with microbiota from an obese human (Turnbaugh 2009). It has also been shown recently by many investigators that the microbiome differs between obese and lean animals and between obese and lean humans (Ley 2005). As yet it is not completely known if intestinal products are the cause or only aggravate NAFLD and NASH. A recent study proposed that the altered microbiome in obesity might produce more ethanol and might thereby contribute to the development of NASH (Zhu 2012). Another recent paper shows that inflammasome or interleukin-18 deficiency enhances the progression of NASH and obesity by inducing microbiome dysbiosis (Henao-Mejia 2012). This dysbiosis-induced inflammation enters into the portal circulation through the influx of toll-like receptor (TLR) 4 and TLR9 agonists and thereby leads to an increase in tumour necrosis factor (TNF) (Moschen 2013). It has also been shown for the first time that the composition of the microbiome and the obese/NASH phenotype can be transmitted to wild-type mice co-housed with genetically deficient mice. This report corroborates that the gut microbiome plays an important role in the development of NASH and obesity, probably via changes in the inflammasome (Henao-Mejia 2012).
Recent metagenome-wide association (MGWAS) studies of gut microbiota showed patients with type 2 diabetes were characterised by a moderate degree of gut microbial dysbiosis, a decrease in the abundance of some universal butyrate-producing bacteria and an increase in various opportunistic pathogens, as well as an enrichment of other microbial functions conferring sulphate reduction and oxidative stress resistance in type 2 diabetes (Qin 2012).
Studies conducted in obese human subjects have confirmed specific changes in the intestinal microbiome, such as a reduction of bacteroidetes and a proportional increase of firmicutes (Ley 2006, Armougom 2009, Santacruz 2010). Moreover, a reduction of bifidobacterium and bacteroides and an increase of staphylococcus, enterobacteriaceae and escherichia coli were detected in overweight compared to normal-weight pregnant women (Santacruz 2010).
A recent MGWAS study analised the human gut microbial composition in 123 non-obese and 169 obese Danish individuals (Le Chatelier 2013). The results showed two groups of individuals that differed by the number of gut microbial genes and thus bacterial richness. Individuals with a low bacterial richness (23% of the population) had more overall visceral adiposity, insulin resistance and dyslipidaemia, and a more pronounced inflammatory phenotype when compared with high bacterial richness individuals. The obese individuals among the lower bacterial richness group also gained more weight over time. Only a few bacterial species were sufficient to distinguish between individuals with high and low bacterial richness, and even between lean and obese subjects. The classifications based on variation in the gut microbiome identified subgroups of individuals in the general white adult population who may be at increased risk of progressing to adiposity-associated co-morbidities (Le Chatelier 2013).
A recent study evaluated the association between gut dysbiosis and severe NAFLD lesions, i.e. non-alcoholic steatohepatitis (NASH) and fibrosis in 57 patients with biopsy-proven NAFLD (Boursier 2015). The taxonomic composition of gut microbiota was determined using 16S ribosomal RNA gene sequencing of stool samples. Thirty patients had F0/1 fibrosis stage at liver biopsy (10 with NASH), and 27 patients had significant F≥2 fibrosis (25 with NASH). Bacteroides abundance was significantly increased in NASH and F≥2 patients, whereas prevotella abundance was decreased. Ruminococcus abundance was significantly higher in F≥2 patients. By multivariate analysis, bacteroides abundance was independently associated with NASH and ruminococcus with F≥2 fibrosis (Boursier 2015). These results suggest that NAFLD severity associates with gut dysbiosis and a shift in metabolic function of the gut microbiota (Boursier 2015). In particular bacteroides may be associated with NASH and ruminococcus with significant fibrosis.
Natural history
The natural history of NAFLD in the general population is not well-defined since most data come from selected patients and tertiary health centres (Dam-Larsen 1996, Lee 1989, Teli 1995). Correspondingly, published mortality and morbidity in hospitalised people with NAFLD are approximately 5 times higher than what is seen in the general population (Matteoni 1999). In the general population, the risk for liver-related death in NAFLD appears to be associated mainly with age, insulin resistance, and histological evidence of hepatic inflammation and fibrosis (Adams 2005). Probably around 10% of NAFLD patients will progress to NASH over a period of 10 years (Figure 1). Cirrhosis later develops in 5–25% of patients with NASH and 30–50% of these patients die from liver-related causes over a 10-year period (McCollough 2005, Matteoni 1999). Cirrhosis in patients with NASH can also decompensate into subacute liver failure, progress to hepatocellular cancer (HCC), and recur after liver transplantation (McCollough 2005). Steatosis alone is reported to have a more benign clinical course, with cirrhosis developing in only 1–3% of patients (Day 2004, Day 2005, McCollough 2005, Matteoni 1999). Patients with NASH and fibrosis also have a significant risk for hepatocellular carcinoma (El-Serag 2004) (Figure 1).
There is no doubt that the incidence and prevalence of NAFLD and NASH are increasing in almost all industrialised countries, sometimes in an epidemic manner. Some recent papers suggest that NASH may soon be the leading cause of cirrhosis, HCC and LTX. There is also growing evidence that HCC in NAFLD and NASH may even develop without cirrhosis. These suggestions would imply that NAFLD related HCC and NAFLD related liver mortality should rapidly increase in the near future. A critical view on recent publications however sheds some doubts on these suggestions.
A retrospective cohort study evaluated trends in the aetiology of HCC among adult recipients of liver transplantation (LTX) in the U.S. from 2002 to 2012 (Wong 2014). During that period 61,868 adults underwent LTX including 10,061 patients with HCC. The proportion of HCV-related HCC increased steadily from 2002 to 2012, and HCV remained the leading aetiology of HCC (43.4% in 2002, 46.3% in 2007, 49.9% in 2012). NASH-related HCC also increased significantly (8.3% in 2002, 10.3% in 2007, 13.5% in 2012) (Wong 2014). Thus, NASH is still only the second leading aetiology of HCC leading to LTX in the U.S. and HCV-related HCC is still more than three-times more prevalent than NAFLD-related HCC.
A PubMed survey analysed original reports published from January 1992 to December 2011 evaluating the association between NAFLD, NASH, cryptogenic cirrhosis presumed to be NASH-related, and the risk of HCC (White 2012). There were 17 cohort studies (3 population based, 9 clinic based, and 5 natural history), 18 case-control and cross-sectional studies, and 26 case series. NAFLD or NASH cohorts with few or no cases of cirrhosis cases had a minimal risk for HCC: the cumulative HCC mortality was only 0–3% for study periods for up to 20 years. Cohorts with NASH and cirrhosis had a consistently higher risk with a cumulative incidence ranging from 2.4% over 7 years to 12.8% over 3 years (White 2012). However, the risk for HCC was substantially lower in these cohorts than for cohorts with hepatitis C-related cirrhosis. This study concluded that there is epidemiologic evidence to support an association between NAFLD or NASH and an increased risk of HCC; such risk seemed to be limited to individuals with cirrhosis (White 2012).
This data is in contrast to another recent multicentre prospective study which assessed the clinical features of patients with NAFLD-related HCC (NAFLD-HCC) and compared them to those of HCV-related HCC (Piscaglia 2015). A total of 756 patients with either NAFLD (n=145) or HCV-related chronic liver disease (n=611) were enrolled in several Italian centres. Cirrhosis was present here in only about 50% of NAFLD-HCC, but in almost all cases of HCV-HCC (Piscaglia 2015).
A recent retrospective study looked at a cohort of Veterans Affairs (VA) patients with the diagnosis of cirrhosis (n = 129,998) or HCC (n = 21,326) from 2001 to 2013 (Beste 2015). Cirrhosis prevalence and mortality, and HCC incidence and mortality increased from 2001 to 2013, driven by HCV, with a much smaller contribution from NAFLD (Beste 2015).
A further study analysed a cohort of 1500 patients who developed HCC between 2005 and 2010 from Veterans Administration (VA) hospitals in the U.S.A. (Mittal 2015a). NAFLD was the underlying risk factor for HCC in 120 patients (8.0%); the annual proportion of NAFLD-related HCC remained relatively stable (7.5%–12.0%). In contrast, the proportion of HCC cases associated with HCV increased from 61.0% in 2005 to 74.9% in 2010 (Mittal 2015a). The proportion of HCC cases associated with only alcohol abuse decreased from 21.9% in 2005 to 15.7% in 2010, and the annual proportion of HCC cases associated with hepatitis B remained relatively low and stable (1.4%–3.5%). A significantly lower proportion of patients with NAFLD-related HCC had cirrhosis (58.3%) compared to patients with alcohol- or HCV-related HCC (72.4% and 85.6%, respectively; P < 0.05). NAFLD was only the third most common risk factor for HCC in the VA population. The proportion of NAFLD-related HCC was relatively stable from 2005 through 2010 (Mittal 2015a).
Further details of this analysis were published separately (Mittal 2015b). This study part looked for evidence of cirrhosis and risk factors for HCC in the VA cohort. Patients without cirrhosis were assigned to categories of level 1 evidence for no cirrhosis (very high probability) or level 2 evidence for no cirrhosis (high probability), which were based on findings from histological analyses, laboratory tests, non-invasive markers of fibrosis, and imaging features. A total of 43 of the 1500 patients with HCC (3%) had level 1 evidence for no cirrhosis, and 151 (10%) had level 2 evidence for no cirrhosis; the remaining 1203 patients (80%) had confirmed cirrhosis. Compared with patients with HCC in presence of cirrhosis, greater proportions of HCC patients without evidence of cirrhosis had metabolic syndrome, NAFLD, or no identifiable risk factors. HCC patients with NAFLD had 5.4-fold risk of having HCC in the absence of cirrhosis, compared to patients with HCV-related HCC. However, only 13% of patients with HCC in the VA system did not appear to have cirrhosis. NAFLD and metabolic syndrome were the main risk factors for HCC in the absence of cirrhosis.
There have been several recent large studies which analysed the long-term outcome of patients with fatty liver disease. They uniformly demonstrated that most patients with NAFLD and also with NASH do not die from liver-related problems but from cardiovascular events (Pisto 2014, Treeprasertsuk 2013, Haflidadottir 2014, Kim 2013).
A large Finnish study analysed a population-based, randomly recruited cohort (Oulu Project Elucidating Risk of Atherosclerosis, OPERA) (Pisto 2014). The study was initiated in 1991 and included 988 middle-aged Finnish participants. Total mortality and hospital events were followed up to 2009 based on the registry of the National Institute for Health and Welfare and the National death registry. The severity of hepatic steatosis was measured by ultrasound and divided into three severity groups. During follow-up between 1991–2009, 13.5% of the participants with non-fatty liver, 24.2% of participants with moderate liver fat content and 29.2% of the participants with severe fatty liver experienced a cardiovascular event (p<0.001). Liver fat content predicted the risk for cardiovascular events even when adjusted for age, gender, smoking, alcohol consumption, LDL cholesterol, BMI, and systolic blood pressure (Pisto 2014).
The Rochester Epidemiology Project also analysed whether the severity of liver fibrosis in NAFLD predicts all-cause mortality, cardiac complications, and/or liver complications (Treeprasertsuk 2013) in a cohort of NAFLD patients during 1980–2000. The NAFLD fibrosis score (NFS) was used to separate NAFLD patients with and without advanced fibrosis. A total of 302 NAFLD patients (mean age: 47 ± 13 year) were included with a follow-up period of 12.0 ± 3.9 years. NFS was < –1.5 at baseline in 181 patients (60%), while NSF was > –1.5 in 121 patients (40%). A total of 39 patients (13%) died during follow-up. The leading causes of death were non-hepatic malignancy (n = 13/39; 33.3%) and coronary heart disease (CHD) (n = 8/39; 20.5%); only 5/39 patients died from liver disease (12.8%). Thirty patients had new-onset CHD, whereas 8 of 30 patients (27%) died from CHD-related causes. In a multivariate analysis, a higher NFS at baseline and the presence of new-onset CHD significantly predicted death. There was a significant, graded relationship between NFS and mortality (Treeprasertsuk 2013). The use of metformin or simvastatin during follow-up was associated with fewer deaths in patients with NAFLD.
A further retrospective study analysed patients who underwent a liver biopsy between 1984–2009 at the National University Hospital of Iceland with NAFLD or AFLD (Haflidadottir 2014). A total of 151 had NAFLD and 94 had AFLD with median survival of 24 years and 20 years, respectively (p >0.05). A total of 10/151 (7%) patients developed cirrhosis in the NAFLD group and 19/94 (20%) in AFLD group (p = 0.03). The most common cause of death in the NAFLD group was cardiovascular disease (48%). In contrast, liver disease was the most common cause of death in the AFLD group (36%), whereas liver-related deaths occurred only in 7% of the NAFLD group. Survival of AFLD patients was significantly shorter compared to the NAFLD patients after adjusting for gender and age at diagnosis (HR 2.16, p = 0.009) (Haflidadottir 2014).
Another large study analysed the long-term impact of NAFLD on mortality using the National Health and Nutrition Examination Survey conducted from 1988–1994 including subsequent follow-up data for mortality through December 31, 2006 (Kim 2013). NAFLD was defined by ultrasound in the absence of other liver diseases. The presence and severity of liver fibrosis in subjects with NAFLD was determined by the NAFLD fibrosis score (NFS), the AST-platelet ratio index (APRI), and the FIB-4 score. Out of 11,154 participants, 34% had NAFLD – the majority (72%) had NFS consistent without significant fibrosis (NFS < −1.455), whereas 3% had a score indicative of advanced fibrosis (NFS > 0.676). After a median follow-up of 14.5 years, NAFLD was not associated with higher mortality [age- and sex-adjusted hazard ratio (HR) 1.05, p>0.1] (Kim 2013). In contrast, there was a progressive increase in mortality with advancing fibrosis scores. Compared to subjects without fibrosis, those with advanced fibrosis had a 69% increase in mortality (HR 1.69 for NFS, HR 1.85 for APRI, HR 1.66 for FIB-4) after adjustment for other known predictors of mortality (Kim 2013). These increases in mortality were almost entirely from cardiovascular causes (HR 3.46 for NFS, HR 2.53 for APRI, HR 2.68 for FIB-4). Thus, ultrasound-diagnosed NAFLD is in general not associated with increased mortality. However, advanced fibrosis as determined by non-invasive fibrosis markers was a significant predictor of mortality, mainly – or even only – from cardiovascular causes (Kim 2013).
Indeed up to 20–50% of all HCC cases may develop in patients with fatty liver disease in the absence of cirrhosis. This percentage is higher when compared with HCV-related HCC. However, almost all publications, also show that HCV-related HCC is still much more frequent than NAFLD-related HCC; the same is also true for the causes of liver transplantation. Recent publications also show that most patients with fatty liver disease die from cardiovascular and not from liver disease (Pisto 2014, Treeprasertsuk 2913, Haflidadottir 2014, Kim 2013). The risk of a cardiovascular death increases with the degree of NAFLD-related liver fibrosis. The worldwide incidence of HCV-related HCC is still increasing while the incidence of NAFLD-related HCC has not increased, but has decreased in the past two decades (Naghavi 2015). The latter unexpected data came from the Global Burden of Disease Study 2013 which recently estimated yearly deaths for 188 countries between 1990 and 2013 (Naghavi 2015). Significant declines were noted for liver cancer due to alcohol use and liver cancer due to other causes (obviously mainly due to fatty liver disease) while significant increases were noted for liver cancer due to hepatitis C. Thus, globally liver cancer due to NAFLD and NASH did not increase but decreased between 1990 and 2013 (Naghavi 2015).
Diagnosis
NAFLD and NASH require valid reporting about alcohol consumption. Since only approximately 10% of Western populations are completely abstinent from alcohol, one needs to set a threshold above which one assumes that alcohol at least contributes to the pathogenic process of NAFLD and NASH. Most authors use a daily alcohol ingestion of 20 g as such a threshold (Figure 2); others use lower values such as 10 g/day or as high as 40 g/day for men.
 Figure 1. Natural history of NASH
Figure 1. Natural history of NASH
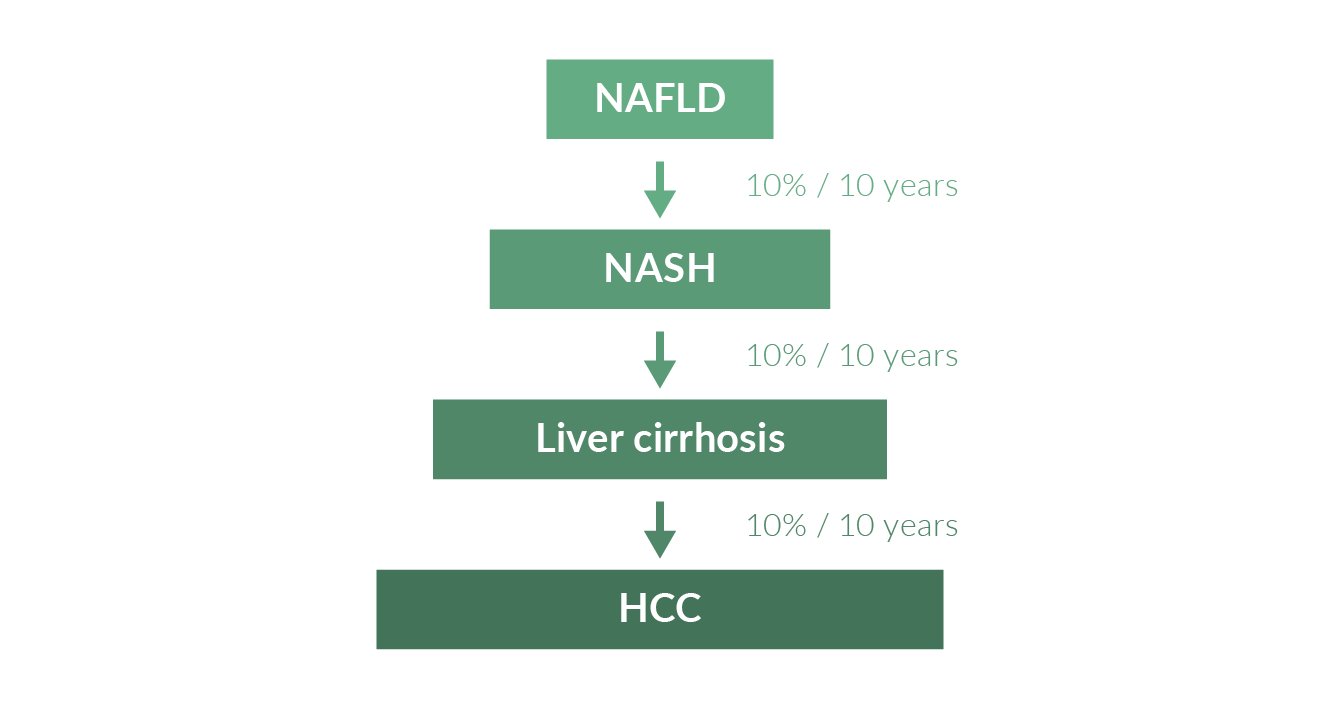 Figure 2. Differentiation of alcoholic liver disease (ASH) and NASH
Figure 2. Differentiation of alcoholic liver disease (ASH) and NASH
The workup of NAFLD and NASH also includes assessment of drug use, exclusion of HBV and HCV infections, haemochromatosis, autoimmune liver disease and, in younger patients, Wilson’s disease. In special groups of patients NASH may be accompanied by drug- and alcohol-induced liver disease and by HCV and HBV infections. The combination of NAFLD/NASH and HCV infection plays a particularly important clinical role because in this situation the rate of liver fibrosis is increased and the success of antiviral therapy is diminished (Ramesh 2004). NASH can be induced by various drugs and toxins including corticosteroids, amiodarone, methotrexate, tetracycline, tamoxifen, and valproate (Pessayre 2002). Thus, one needs to carefully assess the full clinical history of patients. In practice NAFLD is often diagnosed by combining elevated levels of ALT and gamma GT with the sonographic appearance of an increase in echodensity of the liver. However, a considerable number of patients with NAFLD and even with NASH and fibrosis have normal serum liver enzymes (Abrams 2004). Usually ALT is higher than AST unless there is already severe fibrosis or cirrhosis. Fasting serum glucose should be checked in all patients with NAFLD and NASH; one will also often find elevated serum insulin, insulin resistance, and/or diabetes (Table 1).
Table 1. Non-invasive predictors of NASH|
HAIR index (hypertension; ALT >40 U/l; insulin resistance) HAIR score for each patient (0–3) calculated by adding hypertension = 1, ALT 40 IU > 1, and IR index 5.0 > 1. Score ≥2 are 80% sensitive, 89% specific for NASH (Dixon 2001) |
|
BAAT index (BMI >28; Age >50 years; ALT >2x UNL; increased triglycerides) BMI ≥ 28 = 1 point; Age ≥ 50 = 1 point; ALT ≥ 2 × ULN = 1 point; TG ≥ 150 mg/dL = 1 point Score ≤1 has 100% negative predictive value for NASH (Ratziu 2000) |
|
FIB4 score = age (years) x AST [U/L]/(platelets [109/L] x (ALT[U/L]) Using a lower cutoff value of 1.45, a FIB-4 score <1.45 had a negative predictive value of 90% for advanced fibrosis (Ishak fibrosis score 4-6). A FIB4 score >3.25 has a 97% specificity and a positive predictive value of 65% for advanced fibrosis (Sterling 2006; Vallet-Pichard 2007) |
|
BARD score AST/ALT ratio ≥0.8–2 points; a BMI ≥28 – 1 point; and the presence of diabetes – 1 point. Score ranges from 0 to 4 points. Scores equaling 0 or 1 have a high (96%) negative predictive value (NPV) for advanced fibrosis (Harrison 2008) |
|
NFS calculated = -1.675 + 0.037 x age [years] +0.094 x BMI [kg/m2] + 1.13 x impaired fasting glucose or diabetes [yes = 1, no = 0] + 0.99 x AST/ALT ratio– 0.013 x platelet [x 109/L]– 0.66 x albumin g/dL) > 0.676 high probability advanced fibrosis, -1.455 ≤ NFS ≤ 0.676 indeterminate probability of advanced fibrosis, and NFS < -1.455 as low probability of advanced fibrosis (Angulo 2007) |
|
APRI = [AST/upper limit of normal]/ platelet count [109/L] x 100 0.5 for low and 1.5 for high probability of advanced fibrosis (Wai 2003) |
|
The Enhanced Liver Fibrosis (ELF) score includes the three laboratory markers N-terminal peptide of procollagen III (P3NP/PIIINP), hyaluronic acid (HA), and tissue inhibitor of matrix metalloproteinase 1 (TIMP-1). These markers are not in the routine panel in clinical practice. The ELF Score is calculated = 2.278 + 0.851 In (CHA) + 0.751 In (CPIIINP) + 0.394 In (CTIMP-1) A low ELF score (< 7.7) has a high negative predictive value for moderate to severe fibrosis and a high score (> 11.2) has a high positive predictive value for advanced fibrosis or cirrhosis (Lichtinghagen 2013). |
Many authors also recommend to routinely look for metabolic syndrome which is diagnosed when three of the following features are seen (Greenland 2003):
- waist circumference ≥102 cm for men and ≥88 cm for women,
- fasting glucose level ≥6.1 mmol/L,
- triglyceridaemia ≥1.7 mmol/L,
- decreased high-density lipoprotein cholesterol (<1.0 mmol/L in women; <0.9 mmol/L in men)
- hypertension ≥135/80 mmHg.
Ultrasound of the liver has a high sensitivity and specificity (both approaching 90%) for detection of fatty infiltration but does not allow assessment for the presence or degree of inflammation and fibrosis (Davies 1991). Therefore, diagnosis of fat in the liver is easily made by ultrasound but diagnosis of NAFLD or NASH cannot be made without a liver histology. In addition, liver biopsy is still the best way to reliably differentiate NASH from NAFLD (Harrison 2003). Today most pathologists use the Brunt description to score the histological degree of NASH (Brunt 1999) (Table 2).
Table 2. Histological Brunt score (Brunt 1999)| Grade | Steatosis | Ballooning of hepatocytes | Degree of inflammation |
| 1 | <33% | Minimal | Mild |
| 2 | 34–66% | Present | Moderate |
| 3 | >66% | Marked | Portal moderate, lobular moderate |
| Stage | Fibrosis |
| 1 | Perisinusoidal |
| 2 | Perisinusoidal and portal/periportal |
| 3 | Bridging septa |
| 4 | Extensive bridging fibrosis, cirrhosis |
Since NAFLD is a very frequent but also relatively benign disease, one aims to identify risk factors for NASH in order to avoid doing liver biopsies in all NAFLD patients. Risk factors for NASH include older age, excessive obesity, diabetes mellitus, other hepatotoxins, and clinical, laboratory or sonographic signs suggesting severe liver disease. Liver biopsy remains the gold standard for characterising liver histology in patients with NAFLD (Chalasani 2012). However, it is expensive and carries some morbidity and a small mortality risk. Thus, it should be performed in those patients who benefit most from diagnostic, therapeutic and prognostic perspectives. Liver biopsy should be considered in patients with NAFLD who are at increased risk to have steatohepatitis and advanced fibrosis. The presence of metabolic syndrome and the NAFLD Fibrosis Score may be used for identifying patients who are at risk for steatohepatitis and advanced fibrosis (Chalasani 2012). Liver biopsy should also be considered in patients with suspected NAFLD in whom competing etiologies for hepatic steatosis and co-existing chronic liver diseases cannot be excluded without a biopsy (Chalasani 2012).
There has been increasing interest in non-invasive methods to identify liver fibrosis in patients with NAFLD (Gambino 2011; EASL 2016) – these include APRI score, BARD score, HAIR score, BAAT score, NAFLD Fibrosis Score (NFS), Enhanced Liver Fibrosis (ELF) panel, and transient elastography (“Fibroscan”). The NAFLD Fibrosis Score (NFS) is based on six routinely available variables (age, BMI, diabetes, platelet count, albumin, AST/ALT ratio) and is calculated using a published formula (http://nafldscore.com). In a meta-analysis of 13 studies consisting of > 3000 patients, the NFS was useful to predict advanced fibrosis or cirrhosis (Gambino 2011). NFS and FIB-4 score have been externally validated in ethnically different NAFLD populations. According to European guidelines NFS, FIB-4, Enhanced Liver Fibrosis (ELF) and FibroTest predict overall mortality, cardiovascular mortality, and liver-related mortality. NFS predicts incident diabetes, and changes in NFS are associated with mortality. The tests are most useful to distinguish advanced (F3-4) vs. non-advanced fibrosis (F0-2). They not reliably differentiate fibrosis stages F1 from F2 or F0 from F1 (Guha 2008). The negative predictive values (NPVs) for excluding advanced fibrosis are higher than the positive predictive values (PPVs) (Guha 2008; McPherson 2013); thus the EASL recommends to use the non-invasive tests for first-line risk stratification to exclude severe fibrosis (EASL 2016).
Similarly, transient elastography is also an acceptable non-invasive technique to exclude advanced fibrosis and cirrhosis (EASL 2016). A meta-analysis showed that transient elastography has a high sensitivity and specificity for identifying fibrosis in NAFLD patients (Gambino 2011) although it has some problems in individuals with a very high BMI (Chalasani 2012). These problems have partly been solved with a new probe developed for such very obese patients. The technique has been approved by the FDA, but is still not reimbursed in many countries.
The combination of non-invasive scores and transient elastography adds to the diagnostic accuracy of the use of the single procedures and might save diagnostic liver biopsies (EASL 2016). The German NAFLD guidelines recommend to first determine the NFS (Roeb 2015). If its value is lower than -1.455 no other test has to be done since advanced fibrosis is very unlikely. If the NFS is higher than -1.455 transient elastography should be done. If the transient elastography value is higher than 9.6 kPa advanced fibrosis is reliably confirmed and liver biopsy is not neccessary any longer. Liver biopsy is only recommended if transient elastography values are between 7.9 and 9.6 kPa.
Diet, physical exercise and lifestyle recommendations
Table 3. Treatment options for NASH (modified from Chalasani 2012 and Gao 2014)| Moderate weight loss induced by dietary changes and/or physical exercise Abstinence from major alcohol consumption Good control of diabetes mellitus Pioglitazone or vitamin E may be given with caution Liraglutide may be used when diabetes mellitus type 2 and/or obesity are also present Surgery for massive obesity (e.g., gastric bypass surgery) Liver transplantation |
Weight loss generally reduces hepatic steatosis, achieved either by hypocaloric diet alone or in conjunction with increased physical activity (Chalasani 2012) (Table 3). Loss of at least 3-5% of body weight appears necessary to improve steatosis, but a greater weight loss (up to 10%) may be needed to improve necroinflammation. Exercise alone in adults with NAFLD may reduce hepatic steatosis but its ability to improve other aspects of liver histology remains unknown (Chalasani 2012). Several studies have shown that rapid weight loss (very low calorie diet or starving) increases the risk of progression of liver disease and even liver failure (Grattagliano 2000, James 1998, Neuschwander-Tetri 2003). Patients should therefore be educated not to induce rapid weight loss, but to aim at a weight loss of less than 10% of their body weight over 6-12 months (Okita 2001). It is unclear whether special diets are helpful; probably it is more important that the patients simply eat healthy foods like vegetables and fruits, rich in fibre and complex carbohydrates with a low glycemic index; they should avoid meat, saturated fat and products with less complex carbohydrates. Lifestyle modifications should include an increase in physical activity and sports.
There have been several studies, which further evaluated different modes of weight loss due to dietary changes and exercise on fatty liver disease:
A randomised crossover 6-week dietary intervention study examined the effect of a Mediterranean diet (high in monounsaturated fatty acids) on steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease (Ryan 2013). The Mediterranean diet reduced hepatic steatosis and improved insulin sensitivity (Ryan 2013). Another randomised factorial 2 × 2, parallel-group study evaluated the effects of aerobic exercise training and dietary changes on liver fat content in patients with type 2 diabetes (Bozetto 2012): 1) high-carbohydrate/high-fibre/low-glycaemic index diet (CHO/fibre group), 2) high-MUFA diet (MUFA group), 3) high-carbohydrate/high-fibre/low-glycaemic index diet plus physical activity programme (CHO/fibre + Ex group), and 4) high-MUFA diet plus physical activity programme (MUFA + Ex group). Liver fat content decreased more in MUFA (-29%) and MUFA + Ex (-25%) groups than in CHO/fibre (-4%) and CHO/fibre + Ex groups (-6%). Statistics showed a significant effect on liver fat for diet, with no effects for exercise training or diet-exercise interaction. Thus, an isocaloric diet enriched in MUFA compared with a diet higher in carbohydrate and fibre was associated with a reduction of hepatic fat content in type 2 diabetic patients independent of an aerobic training programme and should be considered for the nutritional management of hepatic steatosis in people with type 2 diabetes (Bozetto 2012).
The effects 16 weeks of exercise training were compared with an observation group in a randomised trial (Sullivan 2012). Exercise training resulted in a decrease in hepatic triglyceride content by about 10%, but did not change total body weight or percent body fat. Another randomised controlled trial from Japan evaluated the effect of calorie restriction-induced weight loss with or without aerobic exercise on liver fat content in subjects with visceral adiposity (Yoshimura 2014). Both calorie restriction-induced weight loss without aerobic exercise as well as calorie restriction-induced weight loss with aerobic exercise reduced liver fat content; however, there was no additive effect of exercise training. Another randomised controlled intervention trial compared the effects of 4-months aerobic or resistance training on hepatic fat content in type 2 diabetic subjects with NAFLD (Bacchi 2013). After the training, hepatic fat content was markedly reduced, to a similar extent, in both the aerobic and resistance training groups (mean relative reduction from baseline 32.8% vs. 25.9%). In addition, hepatic steatosis (fat content >5%) disappeared in about one-quarter of the patients in each intervention group. Insulin sensitivity during euglycemic clamp was increased, while total body fat mass and haemoglobin A1c levels were reduced comparably in both intervention groups (Bacchi 2013). Thus, resistance training and aerobic training are equally effective in reducing hepatic fat content among type 2 diabetic patients with NAFLD. One other study examined the effects of aerobic versus resistance exercise without caloric restriction on abdominal adiposity, ectopic fat, and insulin sensitivity in 45 adolescent boys (Lee 2012). Both aerobic and resistance training prevented the significant weight gain observed in control subjects. Compared with controls, total and visceral fat and intrahepatic lipid were reduced in both exercise groups. Both exercise programmes also improved insulin sensitivity (Lee 2012). A further study showed that energy-matched moderate and high intensity exercise training improves nonalcoholic fatty liver disease risk independent of changes in body mass or abdominal adiposity (Winn 2018).
Alcohol and coffee
Several studies suggest a beneficial effect of light alcohol consumption (on average less than one drink per day) on the presence and severity of NAFLD (Suzuki 2007, Dunn 2008, Gunji 2009, Cotrim 2009, Dunn 2009, Moriya 2011). There are no studies reporting the effect of ongoing alcohol consumption on disease severity or natural history of NAFLD or NASH (Chalasani 2012). The effects of light drinking on the cardiovascular system and cancer risks, if any, have not been investigated in individuals with NAFLD. Heavier alcohol consumption is certainly harmful also in obese patients (further literature in Chalasani 2012).
With the results of studies, coffee consumption does not need to be limited and may even have a positive impact on the development of liver fibrosis (Molloy 2012, Birerdinc 2012, Catalano 2010).
Pharmacological treatment
As yet, no drug has been approved by FDA or EMA to treat NASH. However, the 2012 US guidelines (Chalasani 2012) recommend that vitamin E and/or pioglitazone may be given in some patients for treatment of NASH. These recommendations are based in particular on two NIH-sponsored, randomised controlled clinical trials (RCTs) with vitamin E and pioglitazone, the PIVENS and the TONIC trial (Sanyal 2010, Lavine 2011).
Glitazones. The PIVENS study was a large multicentre RCT that randomised 247 non-diabetic patients with NASH to pioglitazone (30 mg/day), vitamin E (800 IU/day) or placebo for 24 months (Sanyal 2010). The primary endpoint was an improvement in >2 NAS points with at least 1 point improvement in hepatocellular ballooning and a 1 point improvement in either the lobular inflammation or steatosis score, and no increase in the fibrosis score. This goal was achieved in 19% of the placebo patients compared to 34% of the pioglitazone-treated patients (p=0.04 vs. placebo) and in 43% of the vitamin E-treated patients (p=0.001 vs. placebo). Because the study consisted of two primary comparisons (pioglitazone vs. placebo and vitamin E vs. placebo), a p-value of 0.025 was considered to be significant a priori. Therefore vitamin E but not pioglitazone met the primary endpoint although there were some histological benefits associated with pioglitazone (Sanyal 2010). It is noteworthy that pioglitazone was associated with a 4.7 kg weight gain compared to placebo (p<0.001). A meta-analysis including 5 RCTs showed that pioglitazone significantly improved steatosis and inflammation, but not fibrosis (Boettcher 2012). Other studies also suggest that pioglitazone improves histological inflammation and fibrosis, and ameliorates cardio-metabolic endpoints in patients not responding to lifestyle intervention (Musso 2012, Chalasani 2012). The other large multicentre RCT, the TONIC study, used the sustained reduction of ALT as the primary endpoint and a change in histology as secondary endpoint (Lavine 2011). The TONIC study compared the efficacy of vitamin E or metformin to placebo for treatment of nonalcoholic fatty liver disease in children and adolescents (8-17 years of age). Although the primary outcome of a reduction of ALT was not different among the three groups, there was a significant improvement in histology (p<0.006) with vitamin E treatment compared to placebo over 96 weeks. In this study, metformin administered at 500 mg twice daily had no effect on aminotransferases and histology (Lavine 2011).
Vitamin E. The US guidelines (Chalasani 2012) state that vitamin E at a daily dose of 800 IU/day improves histology in non-diabetic adults with biopsy-proven NASH and should be considered as first-line treatment. It is also mentioned that vitamin E is not recommended to treat NASH in diabetic patients, NAFLD without liver biopsy, NASH cirrhosis or cryptogenic cirrhosis until further data supporting its effectiveness become available. In addition, the guidelines discuss the controversy as to whether vitamin E increases cancer risks (Chalasani 2012). According to the same guideline (Chalasani 2012) pioglitazone can be used in patients with biopsy-proven NASH. However, it needs be noted that the majority of patients who participated in pioglitazone trials were non-diabetic and that long-term safety and efficacy of pioglitazone in patients with NASH is not established.
Metformin and ursodeoxycholic acid (UDC). Metformin and UDC should not be used for treatment of NASH according to current US guidelines (further literature in Chalasani 2012). Obeticholic acid (OCA) is a semisynthetic derivative of the primary human bile acid chenodeoxycholic acid, the natural agonist of the farnesoid X receptor, which is a nuclear hormone receptor that regulates glucose and lipid metabolism. A double-blind placebo-controlled proof-of-concept study evaluated the effects of OCA on insulin sensitivity in patients with nonalcoholic fatty liver disease and type 2 diabetes mellitus (Mudaliar 2013). OCA treatment for 6 weeks increased insulin sensitivity and reduced markers of liver inflammation and fibrosis in patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease (Mudaliar 2013). However pruritus is a frequent adverse event challenging long term adherence.
Drugs to induce weight loss. In general, all drugs that induce weight loss might be beneficial in NAFLD and NASH, in particular when diet and lifestyle modification do not work (Table 2). Both sibutramine and orlistat have shown to improve some characteristics of NAFLD and NASH such as the sonographic degree of liver steatosis as well as the histological degree of steatosis and fibrosis (Sabuncu 2003, Derosa 2004, Hussein 2007, Harrison 2007). All the latter agents are not approved for use in NASH and NAFLD. Liraglutide is also approved for obsese patients for weight loss, but is not reimbursed for that indication in many countries. The GLP1 analogues however may have a beneficial effect on NAFLD and NASH as well (see following paragraph).
Antioxidants. Antioxidants and cytoprotective agents have also been proposed to treat NAFLD and NASH including vitamin E, vitamin C, vitamin D, pentoxifylline, glutathione, betaine, N-acetylcysteine, S-adenosyl-L-methionine and ursodeoxycholic acid. In a Cochrane analysis, none of these agents showed significant benefit in validated randomised studies (Lirussi 2007). Vitamin D deficiency has been proposed to be involved in the pathogenesis of NASH, and studies proposed that vitamin D supplementation may be useful for treatment of NASH (Barchetta 2012, Roth 2012). There is also one randomised controlled trial suggesting that pentoxifylline might be useful for therapy of non-alcoholic fatty liver disease (Zein 2011). However, larger randomised controlled trials are needed to prove a beneficial role for pentoxifylline in NASH.
Ezetimibe. An open-label randomised controlled clinical trial investigated the efficacy of ezetimibe on NAFLD pathology and insulin sensitivity (Takeshita 2014). The fibrosis stage and ballooning score were significantly improved by ezetimibe. However, ezetimibe treatment significantly increased HbA1c and was associated with a significant increase in hepatic long-chain fatty acids. These findings shed light on previously unrecognised actions of ezetimibe that should be examined further in future studies (Takeshita 2014).
Silybinin. A multicentre, phase 3, double-blind clinical trial assessed the effects of reasil (a silybin phytosome complex consisting of silybin plus phosphatidylcholine, coformulated with vitamin E) versus placebo in patients with histologically documented NAFLD (Loguercio 2012). Patients receiving reasil for 12 months showed significant improvements in liver enzymes, HOMA and liver histology when compared to placebo.
Drug recommendations in guidelines. The Chinese NAFLD guidelines (Gao 2013), the Japanese guidelines (Watanabe 2015), the German guidelines (Roeb 2015) and the informal EASL background information (www.easl.eu) do not support the routine use of any pharmacological agent for NAFLD/NASH patients in view of the available studies. The Chinese guidelines state that there is still insufficient evidence supporting the use of antioxidants and hepatoprotective medications as routine treatment for NAFLD and NASH. Agents included are polyene phosphatidylcholine, vitamin E, silymarin, adenosylmethionine and reduced glutathione. Of all the medications, vitamin E administered at a daily dose of 800 IU/day may be considered as the best liver protectant. According to the Chinese guidelines (Gao 2013) pioglitazone can also be used for the treatment of steatohepatitis in NASH patients. However, there is also a higher rate of congestive heart failure, weight gain and oedema in patients treated with pioglitazone when compared with controls, and the long-term safety and efficacy of pioglitazone in patients with NASH remain uncertain.
Novel pharmacological approaches
Interestingly, NAFLD and NASH are now also the focus of large pharmaceutical companies like Roche, BMS, NGM, MSD and Gilead which are sponsoring studies with interesting new compounds (Ratziu 2012, Stefan 2014). As of April 2018, four drugs are being studies in phase 3 trial and more than 30 in phase 2 trials (www.clinicaltrials.gov).
Inhibition of 11β-hydroxysteroid dehydrogenase type 1. A randomised controlled trial showed that inhibition of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1, also known as HSD11B1) by RO5093151 (a Roche agent) reduced liver-fat content in patients with this disorder (Stefan 2014). This study suggests that targeting of 11β-HSD1 might be a promising approach for the treatment of non-alcoholic fatty liver disease.
Caspase inhibitors. In nonalcoholic steatohepatitis (NASH), the extent of hepatocyte apoptosis correlates with disease severity. Reducing hepatocyte apoptosis with the selective caspase inhibitor GS-9450 (a Gilead agent) has a potential for altering the course of the liver disease. In a phase 3, double-blind study, 124 subjects with biopsy-proven NASH were randomised to placebo or various doses of GS-9450 for 4 weeks (Ratziu 2012). GS-9450 significantly reduced ALT levels in NASH patients (Ratziu 2012). Thus, selective caspase inhibitors may be a promising treatment option for NASH.
The pan-caspase protease inhibitor emricasan has been shown to inhibit apoptosis, inflammation and fibrosis in NASH models (Anstee 2019, Witek 2009). A preliminary report of a phase 2 clinical trial showed significantly decreased serum ALT and cCK18 levels in NAFLD patient (Shiffman 2015). The therapeutic effects of this drug have also been examined in various other fibrotic liver diseases where it has been shown to reduce hepatic venous pressure gradient (HVPG). A phase 2 trial in NASH patients with fibrosis (NCT02686762) is ongoing to evaluate the efficacy and safety of emricasan (10 mg and 100 mg/d for 72 wk) with a special focus on fibrosis without worsening of NASH.
Glucagon-like peptide-1 (GLP-1) analogues. Glucagon-like peptide-1 (GLP-1) analogues reduce hepatic steatosis, concentrations of liver enzymes and insulin resistance in murine models of fatty liver disease. These analogues are licensed for type 2 diabetes and obesity, but their efficacy in patients with NASH was unknown. A multicentre, double-blinded, randomised, placebo-controlled phase 2 trial from four UK centres assessed the effects of s.c. injections of liraglutide (1.8 mg daily) compared with placebo for patients who were overweight and had clinical evidence of NASH (Armstrong 2015). Between 2010-2013, 26 patients were randomly assigned to receive liraglutide and 26 to placebo (Armstrong 2015). Nine (39%) of 23 patients who received liraglutide and underwent end-of-treatment liver biopsy had resolution of NASH compared with two (9%) of 22 such patients in the placebo group (relative risk [RR] 4.3; p=0·019). Two (9%) of 23 patients in the liraglutide group versus eight (36%) of 22 patients in the placebo group had progression of fibrosis (RR 0·2; p=0·04) (Armstrong 2015). Most adverse events were grade 1 (mild) to grade 2 (moderate) in severity, transient, and similar in the two treatment groups for all organ classes and symptoms, with the exception of gastrointestinal disorders in 21 (81%) of 23 patients in the liraglutide group and 17 (65%) of 22 patients in the placebo group, which included diarrhoea (ten [38%] patients in the liraglutide group vs five [19%] in the placebo group), constipation (seven [27%] vs none), and loss of appetite (eight [31%] vs two [8%]). This study was now fully published in the Lancet in 2016 (Armstrong 2016). Liraglutide was safe, well tolerated, and often led to histological resolution of non-alcoholic steatohepatitis, warranting extensive, longer-term studies. Liraglutide has been approved in the US and the EU for treatment of both diabetes mellitus type 2 and for treatment of obesity. Many NASH patients also have diabetes mellitus type 2 and/or obesity. Thus, already today it is possible to treat such patients with liraglutide. In the author’s opinion, liraglutide and similar GLP1-analogues are the most promising pharmacological agents for treatment of NASH and may today already be used when NASH is associated with diabetes or obesity. This personal view is also supported by a meta-analysis (Carbone 2016) which analysed all original studies investigating treatment of adults with NAFLD using GLP-1 analogues. Key outcomes were a change in serum alanine transaminase (ALT), as a marker of liver inflammation, and improvement in disease status measured by imaging or histology. Four studies met all inclusion and exclusion criteria. There were a total of 136 participants with NAFLD and concomitant type 2 diabetes mellitus (T2DM). Meta-analysis (random-effects model) revealed a significant decrease in serum ALT following treatment (mean reduction 14.1 IU/L, P<0.0001). In two studies with imaging and tissue data, treatment was found to significantly reduce steatosis, inflammation, and fibrosis. The significant decrease in a key biochemical marker of hepatic inflammation following treatment with incretin-based therapies, as well as improvements in imaging and histology, suggests these agents may be effective options for managing NAFLD with comorbid type 2 diabetes and/or obesity.
A randomised, placebo-controlled phase 2 trial (NCT0243711) analysed the efficacy and safety of another GLP1-analogue semaglutide in subjects with obesity without type 2 diabetes. Semaglutide induced mean weight losses of -6.0% (0.05 mg) to -13.8% (0.4 mg) (-2.3% with placebo) at week 52. The effect of semaglutide on liver enzymes in subjects with elevated baseline ALT was now evaluated in a post hoc sub-analysis from that trial (Newsome 2018). In subjects with obesity and high ALT, semaglutide at 0.2–0.4 mg once daily s.c. reduced ALT to an extent that was broadly comparable across weight loss categories, and resulted in dose-related ALT normalisation in up to 46% of subjects after 52 weeks. These data suggest a potential role for semaglutide in the treatment of NAFLD with elevated liver enzymes.
Farnesoid X nuclear receptor agonists. The farnesoid X receptor is a nuclear receptor which regulates bile, cholesterol, glucose and lipid metabolism (Pellicciari 2004, Ballestri 2016). These receptors act via multiple pathways inhibiting hepatic lipogenesis, gluconeogenesis, glycogenolysis, maintaining cholesterol balance, and improving insulin sensitivity (Fuchs 2012, Makri 2016). The bile acid derivative 6-ethylchenodeoxycholic acid (obeticholic acid) is a potent activator of the farnesoid X nuclear receptor that reduces liver fat and fibrosis in animal models of fatty liver disease. Obeticholic acid (OCA) is a farnesoid X receptor agonist. It is a synthetic derivative of natural bile acid chenodeoxycholic acid (CDCA); its potency is 100-times greater than that of CDCA. In various animal models, OCA has shown anti-inflammatory and anti-fibrotic properties and also improved insulin resistance and hepatic steatosis (Ali 2015). In an animal model, OCA was shown to reduce hepatic inflammation and fibrosis and also decreased intrahepatic vascular resistance and improved portal hypertension (Verbeke 2016). In an animal model with advanced cirrhosis, treatment with OCA was shown to reduce gut bacterial translocation from 78.3% to 33.3% (p < 0.01) indicating its effect in maintaining intestinal barrier integrity in a further animal model (Ubeda 2016).
A phase 2 multicentre, double-blind, placebo-controlled, parallel group, randomised clinical trial compared treatment with obeticholic acid given orally (25 mg daily) or placebo for 72 weeks at medical U.S. centres in patients with non-cirrhotic NASH (Neuschwander-Tetri 2015). The primary outcome was improvement in liver histology defined as a decrease in NASH activity score by at least 2 points without worsening of fibrosis from baseline to the end of treatment. A planned interim analysis of change in AST at 24 weeks undertaken before end-of-treatment (72 weeks) biopsies supported the decision to continue the trial (relative change in AST -24%, 95% CI -45 to -3). A planned interim analysis of the primary outcome showed improved efficacy of obeticholic acid (p=0.0024) and supported a decision not to do end-of-treatment biopsies and end treatment early in 64 patients, but to continue the trial to obtain the 24-week post-treatment measures (Neuschwander-Tetri 2015). A total of 141 patients were randomly assigned to receive obeticholic acid and 142 to placebo. Fifty (45%) of 110 patients in the obeticholic acid group who were meant to have biopsies at baseline and 72 weeks had improved liver histology compared with 23 (21%) of 109 such patients in the placebo group (RR 1.9; p=0.0002); 33 (23%) of 141 patients in the obeticholic acid developed pruritus compared with nine (6%) of 142 in the placebo group (Neuschwander-Tetri 2015). Also, total cholesterol and LDL cholesterol were significantly increased in the obeticholic acid group when compared with baseline and placebo, while HDL cholesterol was decreased (Neuschwander-Tetri 2015). This lipid profile may be a significant problem using obeticholic acid in NASH since most NASH patients die from cardiovascular and not from liver complications (Pisto 2014, Treeprasertsuk 2013, Haflidadottir 2014, Kim 2013). Nevertheless, a large phase 3 trial is ongoing.
Various further experimental and clinicals trials evaluate other FXR agonists such as GW4064, PX20606, GS-9674 and INT-767. In animal models, these agonists have been shown to improve histological features of NASH such as steatosis and fibrosis (Gege 2014, Carino 2017, Haga 2017, Schwable 2017). Further clinical trials will probably follow.
Polyunsaturated fatty acids. A study by Sanyal et al. (2014) analysed the effect of n-3 polyunsaturated fatty acids (ethyl-eicosapentanoic acid = EPA-E) which are known to reduce insulin resistance, lipogenesis and inflammation. This phase 2b multicentre, prospective, double-blind, randomised, placebo-controlled trial at 37 sites in North America included subjects with NASH and NAFLD activity scores ≥ 4. A total of 243 subjects were randomly assigned to groups given placebo (n = 75), low-dosage EPA-E (1800 mg/d; n = 82), or high-dosage EPA-E (2700 mg/d; n = 86) for 12 months. EPA-E had no significant effects on steatosis, inflammation, ballooning, fibrosis scores, and liver enzymes.
Phosphodiesterase-4 inhibitors. Another trial looked at the effects of the phosphodiesterase-4 inhibitor ASP9831 in patients with NASH after it had shown potent anti-inflammatory and antifibrotic effects in preclinical studies. The phase 2 study evaluated the efficacy and safety of the phosphodiesterase-4 inhibitor ASP9831 in patients with NASH who were assigned randomly to groups given either 50 mg (n = 33) or 100 mg (n = 33) ASP9831 twice daily, or placebo (n = 30) for 12 weeks. After 12 weeks there was no significant change in mean ALT or AST or other biomarkers in any group.
p38 MAPK inhibitors. Chronic inflammation contributes to progression of NAFLD and NASH. The p38 mitogen activated kinases (p38 MAPK) promotes inflammation in the liver (Sabio 2010a, Sabio 2010b, Sabio 2014). Some p38 MAPK isoforms have been shown to contribute to the development of NAFLD and NASH in animal models of fatty liver disease (Gonzalez-Teran 2013, Risco 2012). Human studies have also shown that the hepatic expression of p38 protein is increased in obese individuals with steatosis. Deletion of p38-c and -d in myeloid cells prevented neutrophil migration to the liver and protected these animals against diet-induced steatosis and inflammation (Gonzalez-Teran 2016). Therefore, p38 MAPK may be a promising target for therapy of NAFLD and NASH.
PPAR-α and -δ agonists (elafibranor). The peroxisome proliferator-activated receptors (PPARs) are a family of nuclear receptors that function as transcription factors regulating the expression of various genes thereby regulating e.g. cellular differentiation, development, and metabolism of carbohydrates, lipids, and proteins (Michalik 2006, Dunning 2014). PPAR-α which is mainly expressed in liver regulates many aspects of lipid metabolism. PPAR-δ which is present in various tissues is involved in fatty acid oxidation and insulin sensitivity. In animal models, PPAR has been shown to be hepato-protective by reducing lipid accumulation, inflammation, and fibrosis (Staels 2013, Montagner 2016, Piccinin 2016). In a randomised clinival trial (NCT01694849), a daily dose of 80 or 120 mg elafibranor or placebo was given to non-cirrhotic NASH patients for 52 weeks (Ratziu 2016). The study failed to meet the primary endpoint of resolution of NASH without worsening of fibrosis. However, it was observed that patients with an initial NAS of ≥ 4 treated with 120 mg/d elafribanor showed a significant improvement in liver inflammation. Currently the efficacy of elafibranor is analysed in a phase 3 clinical trial (NCT02704403) in patients with NASH and fibrosis.
NOX-1/4 inhibitors. The NADPH oxidase (NOX) catalyses the production of ROS (Aoyama 2012). In animal models induction of these enzymes by activated hepatic stellate cells promote fibrosis and inflammation (Paik 2011). In a murine model, the NOX-1/4 inhibitor GKT137831 decreased ROS production and fibrotic gene expression and probably thereby reduced hepatic inflammation and fibrosis (Aoyama 2012). Thus, NOX-1/4 inhibitors may have a beneficial effect on liver fibrosis in NAFLD and NASH.
Galectin-3 antagonists. Galectins bind to terminal galactose residues on glycoproteins and are usually expressed in immune cells. Their expression is increased in the presence of inflammation and fibrosis (DiLella 2011, Henderson 2009). Galectin-3 knockout mice show reduced hepatic fibrosis after liver injury. GR-MD-02, a galectin-3 inhibitor, has shown a decrease fibrosis, hepatic steatosis and collagen deposition in various animal models with NASH (Traber 2013). A randomised clinical study analysed the efficacy and safety of the galectin-3 inhibitor GR-MD-02 vs. placebo in patients with NASH and advanced fibrosis (Harrison 2016). GR-MD-02 was safe and well tolerated with evidence of a pharmacodynamic effect. These results provide support for a Phase 2 development programme in advanced fibrosis due to NASH (Harrison 2016).
Acetyl CoA carboxylase inhibitor. Malonyl coenzyme A plays an important role in fatty acid metabolism in maintaining a balance between lipogenesis and lipid oxidation (Foster 2012). It promotes fatty acid synthesis and inhibits β-oxidation of lipids. Malonyl CoA is generated from acetyl CoA and the key enzyme regulating this process is acetyl CoA carboxylase (ACC). Therefore, inhibiting ACC prevents fatty acid synthesis and promotes its oxidation. In a murine model of NAFLD, inhibition of ACC has been shown to decrease hepatic steatosis, lipogenesis and increase insulin sensitivity and fatty acid oxidation (Foster 2012). Administration of the ACC isozyme 1 and 2 inhibitor ND-630 to diet-induced obese rats and Zucker diabetic rats reduced hepatic steatosis, lowered hemoglobin A1C (0.9% reduction), and improved insulin sensitivity (Harriman 2016). Also in a crossover, randomised, double-blind trial, administration of a single dose of NDI-010976 (a highly potent and selective inhibitor of both ACC1 and ACC2) to overweight/obese subjects inhibited de novo lipogenesis in a dose dependent manner (Stiede 2017). Together, all these results suggest its usefulness in treating metabolic syndrome, type 2 diabetes mellitus, and fatty liver disease. Thus, large long term clinical trials in humans are needed.
FGF-21 and FGF-19 analogues. The fibroblast growth factors (FGF) are a family of cell signaling proteins that are involved in a wide variety of processes, most notably as crucial elements for normal development (Burgess 1989). The fibroblast growth factor 21 (FGF-21) is secreted mainly by the liver. It is a starvation-induced peptide hormone with pleiotropic effects whose levels are mainly increased during fasting (Inagaki 2015, Nies 2016). FGF-21 concentrations have been reported to be increased in patients with NAFLD. A reduction of FGF-21 was associated with a metabolic worsening in an animal model of NASH (Liu 2016). FGF21 expression in the liver was reported to be increased in NASH patients (Gallego-Duran 2018). The hepatokine FGF21 is by the liver and regulates sugar intake and preferences for sweet foods via signaling through FGF21 receptors in the suprachiasmatic nucleus and paraventricular nucleus of the hypothalamus as well as the nucleus accumbens and ventral tegmental area (Talukdar 2016, von Holstein-Rathlou 2016, Soberg 2017). Treatment with the FGF-21 analogue BMS-986036 was reported to improve insulin sensitivity, hepatic steatosis and to decrease lipogenesis (Mu 2012). In another animal model of NASH the FGF-21 variant LY240531 was shown to increase fatty acid oxidation by enhancing hepatic mitochondrial oxygen consumption (Liu 2016). Also, various inflammatory markers and AST and ALT levels were reduced, suggesting an attenuation of liver injury (Lee 2016). In a randomised, double-blind, placebo-controlled phase II trial (NCT02413372) (Sanyal 2017) patients with NASH and stage 1-3 fibrosis received the pegylated FGF21 BMS-986036 at 10 mg/day or 20 mg/week for 16 weeks; BMS-986036 dose-dependently reduced the hepatic fat fraction when compared with placebo and was also associated with improvements of adiponectin serum Pro-C3, liver stiffness, lipids, ALT, and AST. The drug was well tolerated without SAEs, discontinuations or deaths (Sanyal 2017). In another phase 2 study of obese patients with type 2 diabetes mellitus, BMS-986036 improved insulin sensitivity and lipids (Charles 2017). An integrated safety analysis of phase 2 studies showed that BMS-986036 was generally safe and well tolerated, supporting further clinical trials to evaluate the safety and efficacy of BMS-986036 in NASH (Halegoua-Demarzi 2018).
The activation FXR in terminal ileum by bile acid induces FGF-19 secretion which then decreases bile acid synthesis and gluconeogenesis (Nies 2016). This process results in the activation of the FGFR4 receptor which has a proliferative effect on hepatocytes with the potential for carcinogenesis (Wu 2010). FGF19 also has important metabolic effects affecting glucose and lipid metabolism when used in experimental mouse models (Tomlinson 2002, Fu 2004, Kir 2011). A randomised, double-blind, placebo-controlled phase 2 study (NCT02443116) assessed the safety and efficacy of the engineered FGF19 analogue NGM282 for the treatment of NASH (Harrison 2018). Patients were randomly assigned (1:1:1) to receive either 3 mg or 6 mg s.c. NGM282 or placebo. Primary endpoint was the absolute change from baseline to week 12 in liver fat content measured by MRI. NGM282 resulted in a rapid and significant reduction of liver fat content with an “acceptable” safety profile in patients with NASH. Further studies of NGM282 are warranted in this patient population (Harrison 2018).
CCR2 and CCR5 inhibitor (cenicriviroc). CCR2 and CCR5 are chemokine receptors which are mainly expressed in immune cells such as monocytes, macrophages, Kupffer cells, natural killer cells, and T cells. They also stimulate hepatic stellate and may thereby initiate fibrosis. Cenicriviroc (CVC) acts as an inhibitor of both the CCR2 and the CCR5 receptor. CVC has been shown to decrease fibrosis and inflammation in various animal models of diet-induced NASH or substance-induced NASH (Lefebvre 2013, Lefebvre 2016, Seki 2009). The ongoing CENTAUR trial (NCT02217475) analyses the efficacy and safety of CVC in NASH patients with fibrosis [255]. Preliminary results after 1 year on study drug have been published (Friedman 2018) and data after 2 years have been shown at the EASL meeting in 2018 (Ratzi 2018). After 1 year of CVC treatment, twice as many subjects achieved improvement in fibrosis without worsening of NASH when compared with placebo (Friedman 2018). A majority of subjects achieving ≥1 stage fibrosis improvement at year 1 maintained that benefit at year 2 with CVC, with greater effect in those with advanced fibrosis (Ratziu 2018).
Inihibtion of acetyl-coenzyme A carboxylase. De novo lipogenesis is an important factor that drives pathogenesis of NASH (Lambert 2014). The rate-limiting step of lipogenesis is catalysed by enzyme acetyl-coenzyme A carboxylase (ACC) (Fullerton 2013). Preclinical and early-phase human studies suggested that an inhibition of ACC may improve steatosis, liver biochemistry, inflammation, fibrosis, and insulin sensitivity (Harriman 2016, Bates 2017, Stiede 2017). More studies analysed the efficacy and safety of the ACC inhibitor GS-0976 in patients with NASH (Loomba 2017, Lawitz 2017). GS-0976 significantly improved liver steatosis and stiffness as well as fibrosis markers in patients with NASH (Loomba 2017, Lawitz 2017).
SCD-1 inhibitors. Stearoyl-CoA desaturase (Δ-9-desaturase) (SCD) catalyses the rate-limiting step in the formation of monounsaturated fatty acids (Paton 2017). SCD-1 is an important metabolic control point. Inhibition of its expression may improve the treatment of a variety of metabolic diseases (Flowers 2017). The SCD1 inhibitor aramchol improved hepatic fat accumulation by decreasing lipogenesis and increasing fatty acid oxidation (Safadi 2014). This drug was found to significantly decrease liver fat content in NAFLD patients when given for 3 months; the effect of the drug on fibrosis was not determined (Safadi 2014). A phase 2 clinical trial of this drug is ongoing to assess the efficacy and safety of aramchol in NASH patients with fibrosis (NCT02279524).
Lysyl oxidase-like 2 inhibitor. Lysyl oxidase-like 2 inhibitors cause cross linkage of collagen and thereby reduce its degradation (Moon 2014). The lysyl oxidase-like 2 enzyme had been shown to promote fibrosis in various liver diseases. Simtuzumab, a monoclonal antibody of lysyl oxidase-like 2, decreased liver fibrosis in animal NASH models (Barry-Hamilton 2017). Two trials examine the efficacy of this drug for decreasing fibrosis and preventing progression to cirrhosis in such patients (NCT01672866, NCT01672879). One of the trials failed to show an effect of simtuzumab on liver fibrosis in NASH patients (Loomba 2017).
Apoptosis signal regulating kinase 1 (ASK1). Apoptosis signal regulating kinase 1 (ASK1) promotes apoptosis and fibrosis induced by hyperglycaemia, TGF-β or ROS. ASK1 is activated in patients with NASH. In animal models of NASH the ASK1 inhibitor GS4997 reduced hepatic steatosis, fibrosis, body weight, fasting blood glucose, insulin resistance, lipogenesis, cholesterol biosynthesis, and plasma AST/ALT levels (Karnik 2014, Karnik 2015, Xiang 2016). A phase 2 study (NCT02466516) evaluated the safety and efficacy of selonsertib, a selective inhibitor of apoptosis signal-regulating kinase 1, alone or in combination with simtuzumab, in patients with NASH and stage 2-3 fibrosis (Loomba 2017). Due to the lack of effect of simtuzumab on histology, selonsertib groups with and without simtuzumab were pooled. After 24 weeks of treatment, the proportion of patients with a ≥ 1 stage reduction of fibrosis in the 18-mg selonsertib group was 13 of 30 (43%; 95% confidence interval, 26-63); in the 6-mg selonsertib group, 8 of 27 (30%; 95% confidence interval, 14-50); and in the simtuzumab-alone group, 2 of 10 (20%; 95% confidence interval, 3-56). Improvement in fibrosis was associated with reductions in MRI liver stiffness, collagen content and lobular inflammation on biopsy, as well as in improvement in serum biomarkers of apoptosis and necrosis. These findings suggest that selonsertib may reduce liver fibrosis in patients with nonalcoholic steatohepatitis and stage 2-3 fibrosis (Loomba 2017).
Pre-clinical data suggest that combinations of an ASK1 inhibitor with an ACC inhibitor or a FXR agonist are more effective than monotherapies. A study evaluated the safety and efficacy of these combinations in subjects with NASH. Patients with NASH received a 12-week treatment with the apoptosis-signal regulating kinase (ASK1) inhibitor (selonsertib) in combination with the acetyl-CoA carboxylase inhibitor (GS-0976) or the farnesoid X receptor agonist (GS-9674) (Lawitz 2018). These combinations were safe and led to improvements in hepatic steatosis, liver biochemistry, and fibrosis markers. Since the responses were similar with mono-therapies, as yet only the safety data are promising for such combinations of drugs.
Sirtuins. Sirtuins (SIRTs) are information regulator proteins. SIRT-1, one member of this protein family, has anti-inflammatory effects and increases insulin sensitivity (Morris 2013). A decreased liver expression of SIRT-1 was observed in an animal model of NAFLD (Colak 2011). Since the SIRT-1 activator resveratrol improved hepatic steatosis and insulin sensitivity (Li 2014), SIRT-1 may be a potential target for treatment of NASH and NAFLD.
Alterations of the intestinal microbiome
Several studies have demonstrated that probiotic strains, in particular those of the lactobacillus and bifidobacterium, exert beneficial effects in subjects with the metabolic syndrome (detailed literature in Ma 2013). Indeed, they seem to promote weight loss, reduce visceral adiposity, improve glucose tolerance and modulate low-grade intestinal inflammation (Ma 2013). In a randomised pilot study patients with histologically proven NASH were randomised to receive probiotics or usual care for 6 months (Wong 2013). Probiotics reduced liver fat and AST level in NASH patients. In another randomised, double-blind, placebo-controlled clinical studies in 52 patients with NAFLD synbiotic supplementation in addition to lifestyle modification was superior to lifestyle modification alone for the treatment of NAFLD, at least partially through attenuation of inflammatory markers (Eslamparast 2014).
Despite encouraging results from these studies and of a meta-analysis evaluating the role of probiotics for the treatment of NAFLD (Ma 2013) other meta-analyses of available randomised clinical trials are more cautious and do not recommend the use of probiotics for the treatment of obesity and fatty liver disease as yet (Millin 2012, Floch 2011). A systematic review analysed randomised clinical trials (RCTs) testing probiotics, prebiotics or both (synbiotics) in adult NAFLD patients (Buss 2014). After the screening process, 9 full-text articles were included in the review, but 6 studies were excluded for methodological problems. Three randomised controlled trials were finally included in the analysis. Patients in these three studies were randomised to receive different formulations of probiotics, synbiotics or placebo. Reductions in aminotransferases were observed in the treated group in 2 of the 3 studies. However, in one study reductions were also detected in the control group. This latest meta-analysis concludes that the current evidence precludes recommendations on the use of pre- and probiotics in clinical practice (Buss 2014). The guidelines also do not recommend such use of pre- and probiotics in clinical practice yet (Chalasani 2012, Gao 2014).
A pilot human study evaluated the effects of infusing intestinal microbiota from lean donors to male recipients with metabolic syndrome on the recipients’ microbiota composition and glucose metabolism. Subjects were assigned randomly to groups that were given small intestinal infusions of allogenic or autologous microbiota. Six weeks after infusion of microbiota from lean donors, insulin sensitivity of recipients increased along with levels of butyrate-producing intestinal microbiota. Intestinal microbiota might be developed as therapeutic agents to increase insulin sensitivity (Vrieze 2012).
Another pilot study explored the role of microbiota in pregnancy characterising faecal bacteria of 91 pregnant women of varying pre-pregnancy BMIs and gestational diabetes status and their infants (Koren 2012). Similarities between infant-mother microbiota increased with children’s age, and the infant microbiota was unaffected by mother’s health status. Gut microbiota changed dramatically from first (T1) to third (T3) trimesters, with an overall increase in proteobacteria and actinobacteria, and reduced richness. T3 stool showed strongest signs of inflammation and energy loss. When transferred to germ-free mice, T3 microbiota induced greater adiposity and insulin resistance compared to T1. These findings indicate that host-microbial interactions that impact host metabolism can occur and may have an impact in pregnancy (Koren 2012).
The composition of the intestinal microbiome determines the efficacy of energy harvest from food. Changes in dietary composition have been associated with changes in the composition of gut microbiota. The capacity to explore the microbiome was markedly improved by the development of metagenomic approach which has led to the first human gut microbial gene catalogue. This approach helps to stratifying individuals by their gut genomic profile into different enterotypes. However, most previous analyses were carried out in non-intervention settings. A pilot study (Cotillard 2013) investigates the temporal relationships between food intake, gut microbiota and metabolic and inflammatory phenotypes, during diet-induced weight-loss and weight-stabilisation interventions in 38 obese and 11 overweight individuals. Individuals with reduced microbial gene richness (40%) showed a low-grade inflammation. Furthermore, dietary intervention improved the low gene richness and clinical phenotypes. Low gene richness may therefore have a potential as an efficacy marker of an intervention (Cotillard 2013).
It has been corroborated that an animal-based diet can rapidly increase the abundance of bile-tolerant micro-organisms (alistipes, bilophila and bacteroides) and may decrease the levels of firmicutes that metabolise dietary plant polysaccharides (roseburia, eubacterium rectale and ruminococcus bromii) (David 2014). Microbial activity mirrored differences between herbivorous and carnivorous mammals. This data demonstrates that the gut microbiome can rapidly respond to altered diet, potentially facilitating the diversity of human dietary lifestyles (David 2014).
In another study a murine model of high-fat diet-induced NAFLD was used to look at the effects of alterations in the intestinal microbiome on NAFLD characteristics (Jian 2014). Mice treated with antibiotics exhibited altered bile acid composition, with an increase in conjugated bile acid metabolites that inhibited intestinal farnesoid X receptor (FXR) signalling. Compared with control mice, animals with intestine-specific FXR disruption had reduced hepatic triglyceride accumulation in response to a high-fat diet. Other parts of this study demonstrated that inhibition of the intestinal FXR/ceramide axis mediates gut microbiota-associated NAFLD development, linking the microbiome, nuclear receptor signalling and NAFLD. This work suggests that inhibition of intestinal FXR is a potential therapeutic target for NAFLD treatment (Jiang 2014).
Surgery for obesity
Bariatric surgery has shown to improve NASH (Liu 2007, de Almeida 2006, Furuya 2007). The US guidelines (Chalasani 2012) state that bariatric surgery is not contraindicated in otherwise eligible obese individuals with NAFLD or NASH.
Until recently there were no randomised studies evaluating bariatric surgery in NAFLD and NASH patients. The first randomised controlled study compared the effects of a step 1 American Heart Association diet plus exercise and intragastric balloon placement to a step 1 American Heart Association diet plus exercise and sham intragastric balloon placement over a period of 6 months (Lee 2012). At 6 months, BMI and the nonalcoholic fatty liver disease activity score were significantly lower in the intragastric balloon placement group when compared with the sham-treated group with an additional trend toward improvement in the steatosis score. There was no change in the median lobular inflammation, hepatocellular ballooning or fibrosis score in either group (Lee 2012). This being the only randomised study, it appears premature to recommend bariatric surgery as a standard option to treat NASH.
A number of long-term studies have been published all of which support the beneficial metabolic value of bariatric surgery some of them with a randomised controlled trial (RCT) design (Adams 2017, Kalinowski 2017, Klebanoff 2017, Pereli 2018, Schauer 2017).
Liver transplantation (LTX) for NASH
LTX is the final option for patients with end-stage liver disease due to cirrhosis and complications of portal hypertension with NASH. Due to the increase in the prevalence of NASH, there is also an increase in LTX due to end-stage liver disease caused by NASH (Burke 2004). Additional reports corroborate that NASH-related LTX has further increased (Wong 2014). However, NASH is still only the second or third leading cause of LTX in the U.S. (for further discussion and references see chapter on Natural History). NASH can recur after LTX, particularly if patients have previously undergone jejunoileal bypass surgery (Kim 1996, Requart 1995, Weston 1998, Contos 2001, Burke 2004). LTX does not cure the metabolic defect that causes NASH.
Follow-up of NAFDL and NASH patients
Patients with NASH cirrhosis should be screened for gastroesophageal varices and for hepatocellular cancer (HCC) (Chalasani 2012) according to current practice guidelines (Garcia-Tsao 2007, Bruix 2011). Although a HCC may occur even in NASH patients without cirrhosis, the relative paucity of such an event does probably not justify to also screen non-cirrhotic NASH patients for HCC. Current evidence does not support to routinely repeat liver biopsy in patients with NAFLD or NASH (Chalasani 2012).
References
Abrams GA, Kunde SS, Lazenby AJ, Clements RH. Portal fibrosis and hepatic steatosis in morbidly obese subjects: a spectrum of nonalcoholic fatty liver disease. Hepatology 2004;40:475-83.
Adams LA, Lymp JF, Sauver SJ, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005;129:113-21.
Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 2005;42:132-8.
Adams TD, Davidson LE, Litwin SE, et al. Weight and metabolic outcomes 12 years after gastric bypass. N Engl J Med 2017;377:1143-1155.
Ali AH, Carey EJ, Lindor KD. Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 2015; 3: 5
Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007;45:846-54.
Anstee QM, Concas D, Kudo H, et al. Impact of pan-caspase inhibition in animal models of established steatosis and nonalcoholic steatohepatitis. J Hepatol 2010; 53: 542-550.
Aoyama T, Paik YH, Watanabe S, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012; 56: 2316-2327.
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2015 Nov 19. pii: S0140-6736(15)00803-X. doi: 10.1016/S0140-6736(15)00803-X. [Epub ahead of print]
Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016 ;387:679-90.
Armougom F, Henry M, Vialettes B, et al. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One 2009; 4: e7125
Bacchi E, Negri C, Targher G, et al. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 Randomized Trial). Hepatology. 2013;58:1287-95.
Baclig MO, Lozano-Kühne JP, Mapua CA, et al. Genetic variation IL48M in patatin-like phospholipase 3 gene and risk of non-alcoholic fatty liver disease among Filipinos. Int J Clin Exp Med 2014;7:2129-36.
Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718-23.
Bahcecioglu IH, Koruk M, Yilmaz O, et al. Demographic and clinicopathological characteristics of nonalcoholic fatty liver disease in the east-southeastern Anatolia regions in Turkey. Med Princ Pract 2006;15:62-8.
Ballestri S, Nascimbeni F, Romagnoli D, Baldelli E, Lonardo A. The Role of Nuclear Receptors in the Pathophysiology, Natural Course, and Drug Treatment of NAFLD in Humans. Adv Ther 2016; 33: 291-319.
Barchetta I, Carotti S, Labbadia G, et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012;56:2180-7.
Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidaselike-2 impedes the development of a pathologic microenvironment. Nat Med 2010; 16: 1009-1017.
Bates J, Hollenback D, Zagorska A, et al. Combination of ASK1 and ACC inhibitors increases efficacy in rodent models of NASH. Program and abstracts of the 2017 Annual Meeting of the American Association for the Study of Liver Diseases; October 20-24, 2017; Washington DC. Abstract 425.
Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005;42:44-52.
Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297-307.
Bellentani S, Tiribelli C, Saccoccio G, et al. Prevalence of chronic liver disease in the general population of northern Italy: the Dionysos Study. Hepatology 1994;20:1442-9.
Beste LA, Leipertz SL, Green PK, et al. Trends in burden of cirrhosis and hepatocellular carcinoma by underlying liver disease in US Veterans, 2001-2013. Gastroenterology 2015;149:1471-1482.
Bonkovsky HL, Jawaid Q, Tortorelli K, et al. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol. 1999;31:421-9.
Boursier J, Mueller O, Barret M, et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2015 Nov 24. doi: 10.1002/hep.28356. [Epub ahead of print]
Bozzetto L, Prinster A, Annuzzi G, et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012;35:1429-35.
Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387-95.
Bruix J, Sherman M. Management of Hepatocellular carcinoma: An update. Hepatology 2011;53:1020-2.
Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467-74.
Burgess WH, Maciag T. The heparin-binding (fibroblast) growth factor family of proteins. Annu Rev Biochem. 1989; 58: 575-606.
Burke A, Lucey MR. Non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and orthotopic liver transplantation. Am J Transplant 2004;4:686-93.
Burza MA, Pirazzi C, Maglio C, et al. PNPLA3 IL48M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig Liver Dis 2012;44:1037-41
Buss C, Valle-Tovo C, Miozzo S, Alves de Mattos A. Probiotics and synbiotics may improve liver aminotransferases levels in non-alcoholic fatty liver disease patients. Ann Hepatol 2014;13:482-8.
Carbone LJ, Angus PW, Yeomans ND. Incretin-based therapies for the treatment of non-alcoholic fatty liver disease: A systematic review and meta-analysis.
J Gastroenterol Hepatol 2016;31:23-31.
Cai T, Dufour JF, Muellhaupt B, et al. Viral genotype-specific role of PNPLA3, PPARG, MTTP, and IL28B in hepatitis C virus-associated steatosis. J Hepatol 2011;55:529-35
Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol 2004;40:578-84.
Carino A, Cipriani S, Marchianò S, et al. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci Rep 2017; 7: 42801.
Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012;58:2005-23.
Charles ED, Neuschwander-Tetri BA, Luo Y, et al. A phase 2 study of BMS-986036 (pegylated FGF21) in obese adults with type 2 diabetes and a high prevalence of fatty liver. Program and abstracts of the 67th Annual Meeting of the American Association for the Study of Liver Diseases; November 11-15, 201; Boston, Massachusetts. Abstract 33.
Chitturi S, Farrell GC, George J. Non-alcoholic steatohepatitis in the Asia-Pacific region: future shock. J Gastroenterol Hepatol 2004;19:368-74.
Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology 2002;122:1649-57
Colak Y, Ozturk O, Senates E, et al. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med Sci Monit 2011; 17: HY5-HY9.
Contos MJ, Cales W, Sterling RK, et al. Development of nonalcoholic fatty liver disease after orthotopic liver transplantation for cryptogenic cirrhosis. Liver Transpl 2001;7:363-73.
Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature 2013;500:585-8.
Cotrim HP, Freitas LA, Alves E, et al. Effects of light-to-moderate alcohol consumption on steatosis and steatohepatitis in severely obese patients. Eur J Gastroenterol Hepatol 2009;21:969-72.
Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142:711-725.
Dam-Larsen S, Franzmann M, Andersen IB, et al. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut 2004;53:750-5.
David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63.
Davies RJ, Saverymuttu SH, Fallowfield M, Joseph AE. Paradoxical lack of ultrasound attenuation with gross fatty change in the liver. Clin Radiol 1991;43:393-6.
Davis JN, Lê KA, Walker RW, et al. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr 2010;92:1522-7
Day CP. The potential role of genes in nonalcoholic fatty liver disease. Clin Liver Dis 2004;8:673-91.
Day CP. Natural history of NAFLD: remarkably benign in the absence of cirrhosis. Gastroenterology 2005;129:375-8.
de Almeida SR, Rocha PR, Sanches MD, et al. Roux-en-Y gastric bypass improves the nonalcoholic steatohepatitis (NASH) of morbid obesity. Obes Surg 2006;16:270-8.
Delahanty LM, Pan Q, Jablonski KA, et al. Genetic predictors of weight loss and weight regain after intensive lifestyle modification, metformin treatment, or standard care in the Diabetes Prevention Program. Diabetes Care 2012;35:363-6.
De Nicola S, Dongiovanni P, Aghemo A, et al. Interaction between PNPLA3 IL48M variant and age at infection in determining fibrosis progression in chronic hepatitis C. PLoS One. 2014;9:e106022.
Derosa G, Cicero AF, Murdolo G, Ciccarelli L, Fogari R. Comparison of metabolic effects of orlistat and sibutramine treatment in Type 2 diabetic obese patients. Diabetes Nutr Metab 2004;17:222-9.
Di Lella S, Sundblad V, Cerliani JP, et al. When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 2011; 50: 7842-7857.
Di Stefano JK, Kingsley C, Craig Wood G, et al. Genome-wide analysis of hepatic lipid content in extreme obesity. Acta Diabetol. 2014 Sep 23. [Epub ahead of print]
Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001;121:91-100.
Dongiovanni P, Donati B, Fares R, et al. PNPLA3 IL48M polymorphism and progressive liver disease. World J Gastroenterol 2013;19:6969-78
Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008;47:1947-54.
Dunn W, Brunt EM, Sanyal AJ, et al. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). Hepatology 2009; 50:390A.
Dunning KR, Anastasi M R, Zhang VJ, et al. Regulation of Fatty Acid Oxidation in Mouse Cumulus-Oocyte Complexes during Maturation and Modulation by PPAR Agonists. PLOS ONE 2014; 9: e87327.
EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease.J Hepatol 2016;64:1388-402.
El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004;126:460-8.
Eslamparast T, Poustchi H, Zamani F, et al. Synbiotic supplementation in nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled pilot study. Am J Clin Nutr 2014;99:535-42.
Falleti E, Fabris C, Cmet S, et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int 2011;31:1137-43.
Fassio E, Alvarez E, Dominguez N, Landeira G, Longo C. Natural history of nonalcoholic steatohepatitis: a longitudinal study of repeat liver biopsies. Hepatology 2004;40:820-6.
Floch MH, Walker WA, Madsen K, et al. Recommendations for probiotic use-2011 update. J Clin Gastroenterol 2011; 45 Suppl: S168-S171
Flowers, Matthew T.; Ntambi, James M. Stearoyl-CoA Desaturase and its Relation to High-Carbohydrate Diets and Obesity. Biochim Biophys Acta 2017; 1791: 85–91.
Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest 2012; 122: 1958-1959.
Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754-1767.
Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature 1998;395:763-70.
Friedrich K, Rupp C, Hov JR, et al. A frequent PNPLA3 variant is a sex specific disease modifier in PSC patients with bile duct stenosis. PLoS One 2013;8:e58734.
Friedrich-Rust M, Schoelzel F, Maier S, et al. Severity of coronary artery disease is associated with non-alcoholic fatty liver disease: A single-blinded prospective mono-center study. PLoS One. 2017;12: e0186720.
Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes”. Endocrinology 2004; 145: 2594–603.
Fuchs M. Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid x receptor as an emerging treatment target. J Lipids 2012; 2012: 934396.
Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649-1654.
Furuya CK Jr, de Oliveira CP, de Mello ES, et al. Effects of bariatric surgery on nonalcoholic fatty liver disease: preliminary findings after 2 years. J Gastroenterol Hepatol. 2007;22:510-4.
Gao X, Fan JG. Diagnosis and management of non-alcoholic fatty liver disease and related metabolic disorders: Consensus statement from the Study Group of Liver and Metabolism, Chinese Society of Endocrinology. J Diabetes 2013;5;406-15.
Gallego-Duran R, Martin F, Ampuero J, et al. Genetic and functional analysis of FGF21 in NAFLD/NASH. EASL 2018, Paris. Abstract: THU-472.
Gambino R, Cassader M, Pagano G, et al. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Annals of Medicine 2011;43:617-49.
Garcia-Tsao G, Sanyal J, Grace ND, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007;102:2086-102.
Gege C, Kinzel O, Steeneck C, Schulz A, Kremoser C. Knocking on FXR’s door: the “hammerhead”-structure series of FXR agonists - amphiphilic isoxazoles with potent in vitro and in vivo activities. Curr Top Med Chem 2014; 14: 2143-2158.
Giudice EM, Grandone A, Cirillo G, et al. The association of PNPLA3 variants with liver enzymes in childhood obesity is driven by the interaction with abdominal fat. PLoS One 2011;6:e27933
George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311-8.
González-Terán B, Cortés JR, Manieri E, et al. Eukaryotic elongation factor 2 controls TNF-α translation in LPS-induced hepatitis. J Clin Invest 2013; 123: 164-178.
González-Terán B, Matesanz N, Nikolic I, et al. p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J 2016; 35: 536-552.
Guha IN, Parkes J, Roderick P, Chattopadhyay D, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease:validating the European Liver Fibrosis Panel and exploring simple markers. Hepatology 2008;47:455–460.
Grattagliano I, Caraceni P, Portincasa P, et al. Adaptation of subcellular glutathione detoxification system to stress conditions in choline-deficient diet induced rat fatty liver. Cell Biol Toxicol 2003;19:355-66.
Grattagliano I, Portincasa P, Palmieri VO, Palasciano G. Managing nonalcoholic fatty liver disease. Recommendations for family physicians. Can Fam Physician 2007;53:857-63.
Gunji T, Matsuhashi N, Sato H, et al. Light and moderate alcohol consumption significantly reduces the prevalence of fatty liver in the Japanese male population. Am J Gastroenterol 2009;104:2189-2195.
Grattagliano I, Vendemiale G, Caraceni P, et al. Starvation impairs antioxidant defense in fatty livers of rats fed a choline-deficient diet. J Nutr 2000;130:2131-6.
Greenland P, Knoll MD, Stamler J, et al. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA 2003;290:891-7.
Haga S, Yimin, Ozaki M. Relevance of FXR-p62/SQSTM1 pathway for survival and protection of mouse hepatocytes and liver, especially with steatosis. BMC Gastroenterol 2017; 17: 9.
Halegoua-Demarzi D, Charles E, Tetri B, et al. An integrated safety analysis of phase 2 studies of BMS-986036, a PEGylated fibroblast growth factor 21 analogue for the treatment of non-alcoholic steatohepatitis. EASL 2018, Paris. Abstract: FRI-490.
Haflidadottir S, Jonasson JG, Norland H et al. Long-term follow-up and liver-related death rate in patients with non-alcoholic and alcoholic related fatty liver disease. BMC Gastroenterol 2014;14:166.
Hamaguchi M, Kojima T, Takeda N, et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med 2005;143:722-8.
Harriman G, Greenwood J, Bhat S, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidaemia in rats. Proc Natl Acad Sci U S A. 2016;113:E1796-E1805.
Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol 2003;98:2042-7
Harrison SA, Oliver D, Arnold HL, et al. Development and validation of a simple NALFD clinical scoring system for identifying patient without advanced disease. Gut 2008;57:1441–47.
Harrison SA, Fincke C, Helinski D, Torgerson S, Hayashi P. A pilot study of orlistat treatment in obese, non-alcoholic steatohepatitis patients. Aliment Pharmacol Ther 2004;20:623-8.
Harrison SA, Rinella M, Abdelmalek MF, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018;391:1174-1185.
Hashimoto T, Fujita T, Usuda N, et al. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty
acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J Biol Chem 1999;274:19, 228-36.
He S, McPhaul C, Li JZ, et al. A sequence variation (IL48M)in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 2010;285:6706-15
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012; 482:179-85
Henderson NC, Sethi T. The regulation of inflammation by galectin-3. Immunol Rev 2009; 230: 160-171.
Hernaez R, McLean J, Lazo M, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third national health and nutrition examination survey. Clin Gastroenterol Hepatol 2013;11:1183-90.
Holzapfel C, Grallert H, Huth C, et al. Genes and lifestyle factors in obesity: results from 12,462 subjects from MONICA/KORA. Int J Obes (Lond) 2010;34:1538-45.
Hotta K, Yoneda M, Hyogo H, et al. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med Genet 2010;11:172-7.
Hussein O, Grosovski M, Schlesinger S, Szvalb S, Assy N. Orlistat reverse fatty infiltration and improves hepatic fibrosis in obese patients with nonalcoholic steatohepatitis (NASH). Dig Dis Sci 2007;52:2512-9.
Inagaki T. Research Perspectives on the Regulation and Physiological Functions of FGF21 and its Association with NAFLD. Front Endocrinol 2015; 6: 147.
James OF, Day CP. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol 1998;29:495-501.
Jiang C, Xie C, Li F, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2014 Dec 15. pii: 76738. [Epub ahead of print
Karnik S, Charlton M, Popov Y, et al. Pharmacological inhibition of apoptosis signal-regulating kinase 1 (ASK1) in a murine model of NASH with pre-existing disease blocks fibrosis, steatosis, and insulin resistance. Hepatology 2014; 60: 570A
Kalinowski P, Paluszkiewicz R, Ziarkiewicz-Wróblewska B, et al. Liver function in patients with Nonalcoholic Fatty Liver Disease Randomized to Roux-en-Y Gastric Bypass Versus Sleeve Gastrectomy: A Secondary Analysis of a Randomized Clinical Trial. Ann Surg 2017;266:738-745.
Karnik S, Charlton MR, Li L, et al. Efficacy of an ASK1 Inhibitor to Reduce Fibrosis and Steatosis in a Murine Model of NASH is Associated with Normalization of Lipids and Hepatic Gene Expression and a Reduction in Serum Biomarkers of Inflammation and Fibrosis. Hepatology 2015; 62: 877A
See comment in PubMed Commons belowKim WR, Poterucha JJ, Porayko MK, Dickson ER, Steers JL, Wiesner RH. Recurrence of nonalcoholic steatohepatitis following liver transplantation. Transplantation 1996;62:1802-5.
Kim D, Kim WR, Kim HJ, Therneau TM. Association between non-invasive fibrosis markers and mortality among adults with non-alcoholic fatty liver disease in the United States. Hepatology 2013: 57: 1357–1365.
Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011; 331: 1621–4.
Kitamoto T, Kitamoto A, Yoneda M, et al. Genomewide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet 2013;132:783-92
Klebanoff MJ, Corey KE, Chhatwal J, et al. Bariatric surgery for nonalcoholic steatohepatitis: A clinical and cost-effectiveness analysis. Hepatology 2017;65:1156-1164
Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012;150:470-80.
Kotronen A, Johansson LE, Johansson LM, et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009;52:1056-60.
Lavine JE, Schwimmer JB, Van Natta ML, et al. for the Nonalcoholic Steatohepatitis Clinical Research Network. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659-68.
Lawitz EJ, Poordad F, Coste A, et al. Acetyl-CoA carboxylase inhibitor GS-0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRI-PDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH. Program and abstracts of 2017 European Association for the Study of the Liver annual meeting; April 19-23, 2017; Amsterdam, The Netherlands. Abstract GS-009.
Lawitz E, Herring R, Younes ZH, et al. Proof of concept study of an apoptosis-signal regulating kinase (ASK1) inhibitor (selonsertib) in combination with an acetyl-CoA carboxylase inhibitor (GS-0976) or a farnesoid X receptor agonist (GS-9674) in NASH. EASL 2018, Paris. Abstract: PS-105.
Lambert JE, Ramos-Roman MA, Browning JD, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726-735.
Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500:541-6.
Lee RG. Nonalcoholic steatohepatitis: a study of 49 patients. Hum Pathol 1989;20:594-8.
Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF? and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 1995;96:2461-2468.
Lee S, Bacha F, Hannon T, et al. Effects of aerobic versus resistance exercise without caloric restriction on abdominal fat, intrahepatic lipid, and insulin sensitivity in obese adolescent boys: a randomized, controlled trial. Diabetes 2012;61:2787-95.
Lee YM, Low HC, Lim LG, et al. Intragastric balloon significantly improves nonalcoholic fatty liver disease activity score in obese patients with non-alcoholic steatohepatitis: a pilot study. Gastrointest Endosc 2012;76:756-60.
Lefebvre DR, Hashiguchi T, Jenkins H, et al. Anti-Fibrotic and Anti-Inflammatory Activity of the Dual CCR2 and CCR5 Antagonist Cenicriviroc in a Mouse Model of NASH. Hepatology 2013; 58: 221A-222A.
Lefebvre E, Moyle G, Reshef R, et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS One 2016; 11: e0158156
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005; 102:11070-5.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology:human gut microbes associated with obesity. Nature 2006;444:1022-3
Lee JH, Kang YE, Chang JY, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res 2016; 8: 4750-4763
Li L, Hai J, Li Z, et al. Resveratrol modulates autophagy and NF-κB activity in a murine model for treating non-alcoholic fatty liver disease. Food Chem Toxicol 2014; 63: 166-173
Lichtinghagen R, Pietsch D, Bantel H, et al. The Enhanced Liver Fibrosis (ELF) score: normal values, influence factors and proposed cut-off values. J Hepatol 2013;59:236-42.
Lin YC, Chang PF, Hu FC, et al. A common variant in the PNPLA3 gene is a risk factor for non-alcoholic fatty liver disease in obese Taiwanese children. J Pediatr 2011;158:740-4
Lirussi F, Azzalini L, Orando S, Orlando R, Angelico F. Antioxidant supplements for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst Rev 2007;24:CD004996.
Liu X, Lazenby AJ, Clements RH, Jhala N, Abrams GA. Resolution of nonalcoholic steatohepatits after gastric bypass surgery. Obes Surg 2007;17:486-92.
Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014:30;5:4309.
Liu X, Zhang P, Martin RC, et al. Lack of fibroblast growth factor 21 accelerates metabolic liver injury characterized by steatohepatities in mice. Am J Cancer Res 2016; 6: 1011-1025.
Loguercio C, De Girolamo V, de Sio I, et al. Non-alcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J Hepatol 2001;35:568-74.
Loguercio C, Andreone P, Brisc C, et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: a randomized controlled trial. Free Radic Biol Med 2012;52:1658-65.
Loomba R, Kayali Z, Noureddin M, et al. Acetyl-CoA carboxylase (ACC) inhibitor GS-0976 leads to significant improvements in MRI-PDFF in a phase 2, randomized, placebo-controlled trial of patients with NASH. Program and abstracts of the 2017 Annual Meeting of the American Association for the Study of Liver Diseases; October 20-24, 2017; Washington DC. Abstract LB-9.
Loomba R, Lawitz E, Mantry PS, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2017 Sep 11. doi: 10.1002/hep.29514.
Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980;55:434-8.
Ma YY, Li L, Yu CH, et al. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol 2013; 19: 6911-6918
Makri E, Cholongitas E, Tziomalos K. Emerging role of obeticholic acid in the management of nonalcoholic fatty liver disease. World J Gastroenterol 2016; 22: 9039-9043.
Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001;50:1844-50.
Marchesini G, Brizi M, Morselli-Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 1999;107:450-5.
Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003;37:917-23.
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullogh AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999;116:1413-9.
Marra F. NASH: are genes blowing the hits? J Hepatol 2004;40:853-6
McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol 2006;40:1:S17-S29.
McCullough AJ. The epidemiology and risk factors of NASH. In: Farrell GC, George J, Hall P, McCullough AJ eds. Fatty liver disease: NASH and related disorders. Malden, MA: Blackwell, 2005:23-37.
McPherson S, Anstee QM, Henderson E, Day CP, Burt AD. Are simple noninvasive scoring systems for fibrosis reliable in patients with NAFLD and normal ALT levels? Eur J Gastroenterol Hepatol 2013;25:652-658.
Michalik L, Auwerx J, Berger JP, et al. Peroxisome proliferator-activated receptors. Pharmacol. Rev 2006; 58: 726-41.
Million M, Angelakis E, Paul M, et al. Comparative meta-analysis of the effect of Lactobacillus species on weight gain in humans and animals. Microb Pathog 2012; 53: 100-108
Mittal S, Sada YH, El-Serag HB, et al. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin Gastroenterol Hepatol 2015a;13:594-601.
Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol 2015b Jul 18. pii: S1542-3565(15)00978-7. [Epub ahead of print]
Montagner A, Polizzi A, Fouché E, et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016; 65:1202-1214.
Moon HJ, Finney J, Ronnebaum T, Mure M. Human lysyl oxidase-like 2. Bioorg Chem 2014; 57: 231-241
Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med 2013; 56: 133-171.
Moriya A, Iwasaki Y, Ohguchi Set al. Alcohol consumption appears to protect against non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2011;33:378-88.
Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab 2013;24:537-45.
Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013;145:574-82.
Mu J, Pinkstaff J, Li Z, et al. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes 2012; 61: 505-512.
Müller T, Buch S, Berg T, et al. Distinct, alcohol-modulated effects of PNPLA3 genotype on progression of chronic hepatitis C. J Hepatol 2011;55:732-3.
Naghavi M, Wang H, Lozano R, et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;385:117-71.
Nault JC, Nahon P. Genetic predisposition to hepatocellular carcinoma in alcoholic cirrhosis: The NCAN-PNPLA3-lipid connection? J Hepatol 2014;61:971-2.
Newsome P, Harrison S, Rasmussen S, et al. The effect of semaglutide on liver enzymes in subjects with obesity and elevated alanine aminotransferase: Data from a randomised Phase 2 trial. EASL 2018, Paris. Abstract: FRI-483.
Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology 2003;37:1202-19.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956-65.
Nies VJ, Sancar G, Liu W, et al. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front Endocrinol (Lausanne) 2016; 6: 193.
Nischalke HD, Lutz P, Kramer B, et al. A common polymorphism in the NCAN gene is associated with hepatocellular carcinoma in alcoholic liver disease. J Hepatol 2014;61:1073-9.
Nobili V, Svegliati-Baroni G, Alisi A, et al. A 360-degree overview of paediatric NAFLD: recent insights. J Hepatol 2013;58:1218-29
Okita M, Hayashi M, Sasagawa T, et al. Effect of a moderately energy-restricted diet on obese patients with fatty liver. Nutrition 2001;17:542-7.
Peng XE, Wu YL, Lin SW, et al. Genetic variants in PNPLA3 and risk of non-alcoholic fatty liver disease in a Han Chinese population. PLoS One 2012;7:e50256
Pessayre D, Berson A, Fromenty B, Mansouri A. Mitochondria in steatohepatitis. Semin Liver Dis 2001;21:57-69.
Piscaglia F, Svegliati-Baroni G, Barchetti A, et al. Clinical patterns of hepatocellular carcinoma (HCC) in non alcoholic fatty liver disease (NAFLD): A multicenter prospective study. Hepatology. 2015 Nov 24. doi: 10.1002/hep.28368. [Epub ahead of print]
Pisto P, Santaniemi M, Bloigu R, et al. Fatty liver predicts the risk for cardiovascular events in middle-aged population: a population-based cohort study. BMJ Open. 2014 Mar 20;4:e004973.
Ploeg RJ, D’Alessandro AM, Knechtle SJ, Stegall MD, Pirsch JD, Hoffmann RM, et al. Risk factors for primary dysfunction after liver transplantation-a multivariate analysis. Transplantation 1993;55:807-13.
Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55-60.
Paik YH, Iwaisako K, Seki E, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011; 53: 1730-1741.
Paton, Chad M.; Ntambi, James M. Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metabol 2017; 297: E28–E37.
Pellicciari R, Costantino G, Camaioni E, et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem 2004; 47: 4559-4569.
Peterli R, Wölnerhanssen BK, Vetter D, et al. Laparoscopic Sleeve Gastrectomy Versus Roux-Y-Gastric Bypass for Morbid Obesity-3-Year Outcomes of the Prospective Randomized Swiss Multicenter Bypass Or Sleeve Study (SM-BOSS). Ann Surg 2017;265:466-47.
Piccinin E, Moschetta A. Hepatic-specific PPARα-FGF21 action in NAFLD. Gut 2016; 65: 1075-1076.
Polyzos SA, Aronis KN3, Kountouras J, et al Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 2016;59:30-43.
Ramesh S, Sanyal AJ. Hepatitis C and nonalcoholic fatty liver disease. Semin Liver Dis 2004;24:399-413.
Ratziu, et al. Liver Fibrosis in Overweight Patients. Gastroenterology 2000;118:1117-23.
Ratziu V, Sheikh MY, Sanyal AJ, et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012;55:419-28.
Ratziu V, Bedossa P, Francque SM, Larrey D, Aithal GP, Serfaty L, et al. Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2014;12:1724-30.
Ratziu V, Harrison SA, Francque S, et al. GOLDEN-505 Investigator Study Group. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016; 150: 1147-1159.
Ratziu V, Sanyal A, Francque S, et al. Cenicriviroc treatment for adults with non-alcoholic steatohepatitis: Year 2 analysis of the Phase 2b CENTAUR study GS-002. EASL 2018, Paris. Abstract: GS002.
Requarth JA, Burchard KW, Colacchio TA, et al. Long-term morbidity following jejunoileal bypass: the continuing potential need for surgical reversal. Arch Surg 1995;130:318-25.
Risco A, del Fresno C, Mambol A, et al. p38γ and p38δ kinases regulate the toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc Natl Acad Sci USA 2012; 109: 11200-11205.
Roeb E, Steffen HM, Bantel H, et al. S2k Guideline non-alcoholic fatty liver disease. Z Gastroenterol 2015;53:668-723.
Rosenberg WM, Voelker M, Thiel R, et al. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology 2004;127:1704-13.
Roth CL, Elfers CT, Figlewicz DP, et al. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology 2012;55:1103-11. doi: 10.1002/hep.24737. Epub 2012 Feb 15.
Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to non-alcoholic fatty liver disease. Nat Genet 2008;40:1461-5.
Romeo S, Sentinelli F, Dash S, et al. Morbid obesity exposes the association between PNPLA3 IL48M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond) 2010a;34:190-4.
Romeo S, Sentinelli F, Cambuli VM, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol 2010b;53:335-8.
Rotman Y, Koh C, Zmuda JM, et al. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894-903.
Ryan MC, Itsiopoulos C, Thodis T, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol 2013 ;59:138-43.
Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci 2010a; 35: 490-496.
Sabio G, Kennedy NJ, Cavanagh-Kyros J, et al. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol 2010b; 30: 106-115.
Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol 2014; 26: 237-245.
Sabuncu T, Nazligul Y, Karaoglanoglu M, Ucar E, Kilic FB. The effects of sibutramine and orlistat on the ultrasonographic findings, insulin resistance and liver enzyme levels in obese patients with non-alcoholic steatohepatitis. Rom J Gastroenterol 2003;12:189-92.
Safadi R, Konikoff FM, Mahamid M, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2014;122085-91.e1.
Salminen P, Helmiö M, Ovaska J, et. Effect of Laparoscopic Sleeve Gastrectomy vs Laparoscopic Roux-en-Y Gastric Bypass on Weight Loss at 5 Years Among Patients With Morbid Obesity: The SLEEVEPASS Randomized Clinical Trial. JAMA 2018;319:241-254.
Santacruz A, Collado MC, García-Valdés L, et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 2010; 104: 83-92.
Santoro N, Savoye M, Kim G, et al. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS One 2012a;7:e37827
Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory Protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012b;55:781-9.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120:1183-92.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675-85.
Sanyal AJ, Abdelmalek MF, Suzuki A, et al. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 2014;147:377-84.
Sanyal A, Charles ED, Neuschwander-Tetri B, et al. BMS-986036 (PEGylated FGF21) in patients with non-alcoholic steatohepatitis: a phase 2 study. Program and abstracts of the 68th Annual Meeting of the American Association for the Study of Liver Diseases; October 20-24, 2017; Washington, DC. Abstract 182.
Schauer PR,, Bhatt DL, Kirwan JP, et al. Bariatric Surgery versus Intensive Medical Therapy for Diabetes - 5-Year Outcomes. N Engl J Med 2017;376:641-651.
Schwabl P, Hambruch E, Seeland BA, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol 2017; 66: 724-733.
Schwimmer JB, McGreal N, Deutsch R, Finegold MJ, Lavine JE. Influence of gender, race, and ethnicity on suspected fatty liver in obese adolescents. Paediatrics 2005 115:561-5.
Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009; 50: 185-197.
Sevastianova K, Santos A, Kotronen A, et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am J Clin Nutr 2012;96:727-34 .
Shen JH, Li YL, Li D, Wang NN, Jing L, Huang YH. The rs738409 (IL48M) variant of thePNPLA3 gene and cirrhosis: A meta-analysis. J Lipid Res 2014 2014;M048777.
Shang X, Song J, Liu F, et al. Association Study of Seven GWAS-Identified Common Variants with Nonalcoholic Fatty Liver Disease in Chinese Children. J Pediatr Gastroenterol Nutr. 2014 Dec 16. [Epub ahead of print]
Shiffman M, Freilich B, Vuppalanchi R, et al. LP37: A placebo-controlled, multicenter, double-blind, randomised trial of emricasan in subjects with non-alcoholic fatty liver disease (NAFLD) and raised transaminases. J Hepatol 2015; 62: S282.
Søberg S, Sandholt CH, Jespersen NZ, et al. FGF21 Is a Sugar-Induced Hormone Associated with Sweet Intake and Preference in Humans. Cell Metabolism 2017 25: 1045–1053.
Sookoian S, Castaño GO, Burgueño AL, et al. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111-6.
Sookoian S, Pirola CJ. Meta-analysis of the influence of IL48M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883-94.
Sookoian S, Castaño GO, Scian R, et al. Genetic variation in TM6SF2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology. 2014 Oct 10. doi: 10.1002/hep.27556.
Speliotes EK, Butler JL, Palmer CD, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010;52:904-12.
Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324.
Staels B, Rubenstrunk A, Noel B, et al. Hepatoprotective effects of the dual peroxisome proliferator activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013; 58: 1941-1952.
Stefan N, Ramsauer M, Jordan P, et al. Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2014;2:406-16.
Sterling RK, Lissen E, Clumeck N, et. al. Development of a simple noninvasive index to predict significant fibrosis patients with HCV/HIV co-infection. Hepatology 2006;43:1317-1325.
Stiede K, Miao W, Blanchette HS, et al. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: a randomized, double-blind, crossover study. Hepatology. 2017;66:324-334.
Sullivan S, Kirk EP, Mittendorfer B, et al. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology 2012;55:1738-45.
Suzuki A, Angulo P, St. Sauver J, Muto et al. Light to moderate alcohol consumption is associated with lower frequency of hypertransaminasemia. Am J Gastroenterol 2007;102:1912-9.
Suzuki A, Angulo P, Lymp J, Li D, Satomura S, Lindor K. Hyaluronic acid, an accurate serum marker for severe hepatic fibrosis in patients with non-alcoholic fatty liver disease. Liver Int 2005;25:779-86.
Takeshita Y, Takamura T, Honda M, et al. The effects of ezetimibe on non-alcoholic fatty liver disease and glucose metabolism: a randomised controlled trial. Diabetologia 2014;57:878-90.
Talukdar S, Owen BM, Song P, et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metabolism 2016: 23: 344–9.
Tomlinson E, Fu L, John L, Hultgren B, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002; 143: 1741–7.
Tong J, Guo J, Hu J, et al. Correlation between patatin-like phospholipase domain-containing protein 3 gene polymorphisms and liver cirrhosis in a chinese han population with chronic hepatitis B. Hepat Mon 2014;14:e18943.
Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS One 2013; 8: e83481.
Treeprasertsuk S, Björnsson E, Enders F, et al. NAFLD fibrosis score: a prognostic predictor for mortality and liver complications among NAFLD patients. World J Gastroenterol 2013;19:1219-29.
Trépo E, Pradat P, Potthoff A, et al. Impact of patatin-like phospholipase-3 (rs738409 C > G) polymorphism on fibrosis progression and steatosis in chronic hepatitis C. Hepatology 2011;54:60-9-
Trepo E, Guyot E, Ganne-Carrie N, et al. PNPLA3 (rs738409 C > G) is a common risk variant associated with hepatocellular carcinoma in alcoholic cirrhosis. Hepatology 2012;55:1307-8.
Trepo E, Nahon P, Bontempi G, et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014;59:2170–7.
Tuyama AC, Chang CY. Non-alcoholic fatty liver disease. J Diabetes 2012;4:266-80.
Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology 1995;22:1714-9.
Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027-31.
Turnbaugh PJ, Ridaura VK, Faith JJ, et al. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009;1:6ra14.
Úbeda M, Lario M, Muñoz L, Borrero MJ, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol 2016; 64: 1049-1057.
Valenti L, Al-Serri A, Daly AK, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin IL48M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209-17.
Valenti L, Rumi M, Galmozzi E, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011; 53:791-799.
Valenti L, Maggioni P, Piperno A, et al. Patatin-like phospholipase domain containing-3 gene I148M polymorphism, steatosis, and liver damage in hereditary haemochromatosis. World J Gastroenterol 2012;18:2813-20
Valenti L, Alisi A, Nobili V. Unraveling the genetics of fatty liver in obese children: additive effect of P446L GCKR and IL48M PNPLA3 polymorphisms. Hepatology 2012;55:661-3.
Valenti L, Dongiovanni P, Ginanni C, et al. PNPLA3 I148M variant and hepatocellular carcinoma: a common genetic variant for a rare disease. Dig Liver Dis 2013;45:619-24.
Vallet-Pichard A, Mallet V, Nalpas B, et al. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. comparison with liver biopsy and fibrotest. Hepatology. 2007;46:32-6.
Vendemiale G, Grattagliano I, Caraceni P, et al. Mitochondrial oxidative injury and energy metabolism alteration in rat fatty liver: effect of the nutritional status. Hepatology 2001;33:808-15.
Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 2016; 6: 33453.
Viganò M, Valenti L, Lampertico P, et al. Patatin-like phospholipase domain-containing 3 I148M affects liver steatosis in patients with chronic hepatitis B. Hepatology 2013;58:1245-52.
Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012;143:913-6.
von Holstein-Rathlou S, BonDurant LD, Peltekian L, et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metabolism 2016: 23: 335-43.
Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38:518–26.
Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 1990;12:1106-10.
Watanabe S, Hashimoto E, Ikejima K, et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatol Res 2015;45:363-77.
Weston S, Charlton MR, Lindor KD. Liver transplantation for nonalcoholic steatohepatitis. Gastroenterology 1998;114:A1364.
Weston SR, Leyden W, Murphy R, et al. Racial and ethnic distribution of nonalcoholic fatty liver in persons with newly diagnosed chronic liver disease. Hepatology 2005;41:372-9.
White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012;10:1342-1359.
Winn NC, Liu Y, Rector RS, Parks EJ, Ibdah JA, Kanaley JA. Energy-matched moderate and high intensity exercise training improves nonalcoholic fatty liver disease risk independent of changes in body mass or abdominal adiposity - A randomized trial. Metabolism 2018;78:128-140.
Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009; 50: 1421-1430.
Wong VW, Won GL, Chim AM, et al. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol 2013;12:256-62.
Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014;59:2188-95.
Wu X, Ge H, Lemon B, et al. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem 2010; 285: 5165-5170.
Xiang M, Wang PX, Wang AB, et al. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J Hepatol 2016; 64: 1365-1377.
Yuan X, Waterworth D, Perry JR, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 2008;83:520-8.
Yoshimura E, Kumahara H, Tobina T, et al. Lifestyle intervention involving calorie restriction with or without aerobic exercise training improves liver fat in adults with visceral adiposity. J Obes 2014;2014:197216. Epub 2014 Apr 17.
Younossi ZM, Gramlich T, Bacon BR, et al. Hepatic iron and nonalcoholic fatty liver disease. Hepatology 1999;30:847-50.
Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54:1610-9.
hu L, Baker SS, Gill C, et al. Characterization of the gut microbiome in non-alcoholicsteatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013;57:601-9.