
5. Hepatitis E
Sven Pischke, Katja Dinkelborg, Patrick Behrendt
Epidemiology
Hepatitis E is an inflammatory liver disease caused by infection with the hepatitis E virus (HEV): It has been described to be endemic in many tropical countries with reduced sanitary conditions in the 1980ies. For more than two decades, it was considered to be a travel-associated, acute, self-limiting liver disease that only causes fulminant hepatic failure in specific, high-risk groups (Pischke 2013b). More recently, HEV infection was estimated to cause approximately 56, 000 deaths each year worldwide (WHO 2014). Within the last two decades sporadic cases of HEV infections have emerged also in industrialised countries, mostly caused by HEV genotype 3, which are mostly caused by zoonotic transmission (Wedemeyer 2012, Pischke 2013).
Today we know that HEV infections occur worldwide but the genotypes are distributed differently (Figure 1). HEV is classified into the family of Hepeviridae and belongs to the subfamily of Orthohepevirinae which includes four species. Until recently, infections to humans have only been described from the species A with 8 genotypes. Recently, transmission of rat-HEV (Orthohepevirinae C) from rodents to humans have also been described (Rivero-Juarez 2022).
The vast majority of cases of hepatitis E worldwide is caused by HEV genotype 1 and 2 infections transmitted by the fecal-oral route. These are causative for outbreaks and are transmitted by contaminated drinking water. For examplean outbreak in south sudan with a case-fatality ratio of 5.5% occurred in the spring of 2023 (WHO 2023) In contrast, genotypes 3 and 4 are mostly transmitted zoonotically in industrialised countries (figure 1). Both direct contact with HEV-infected domestic animals and foodborne transmission are possible routes of transmission (Wedemeyer 2012). Commercial food products such as pig meat may be contaminated with HEV as shown in studies from the Netherlands, France and Germany (Colson 2010, Melenhorst 2007, Wenzel 2011). Meat should be cooked at above 70°C to prevent foodborne HEV infections (Emerson 2005). In the last few years, an increasing frequency of diagnosed cases of HEV infections has been reported from various industrialised countries (Wedemeyer 2012). The presence of HEV RNA in urban sewage samples from Spain, the US and France has been shown, suggesting that HEV may be more prevalent in industrialised countries than previously assumed (Clemente-Casares 2003). In each of these three countries it was possible to discover HEV contamination in sewage samples in a notably high frequency. These findings may partially explain the huge gap between seroprevalence rates and the rather low numbers of diagnosed and reported cases of acute hepatitis E in western countries. The mismatch between high seroprevalence rates and the low number of symptomatic cases has also been investigated in a recent study from Egypt. 919 anti-HEV seronegative individuals from rural Egypt were followed and, interestingly, 3.7% (n=34) of these individuals seroconverted to anti-HEV within 11 months of follow up (Stoszek 2006). However, none of these 34 individuals suffered from symptomatic hepatitis E. This finding corresponds with data from a recently published large vaccine study performed in China where very few of the patients in the placebo group who seroconverted during a follow-up period developed symptomatic acute hepatitis E (Zhu 2010). Overall, these data suggest that far less than 5% of all contacts with HEV lead to symptomatic hepatitis E (Wedemeyer 2011).
Genotypes 5-8 occur in animals (wild boars, camels), but play only a minor role in humans, with only a few published cases. For example an HEV genotype 7 infection transmitted by camel meat leading to chronic hepatitis E in a liver transplant recipient has been demonstrated (Lee 2015). Although this is surely of limited relevance in European countries and the USA, it highlights a novel mode of transmission in Arabian countries. In these Islamic countries, HEV genotype 3 and 4 infections originating from pigs certainly play a minor role.
Furthermore the variant "rat-HEV" (HEV-C) has attracted increased attention in recent years, as cases of human infections caused by this variant have been diagnosed in Hong Kong, Spain and France (Rivero-Juarez 2022). This HEV variant differs genetically from the other versions to such an extent, that the conventional commercial HEV PCR tests do not detect it and specially designed primers are needed. This especially poses a threat to being underdiagnosed in the clinic and in the case of severe hepatitis of an unknown cause, HEV-C infection should be considered.
Reference sequences have been published to facilitate communication between researchers and enable improved classification of HEV strains (Smith 2016).
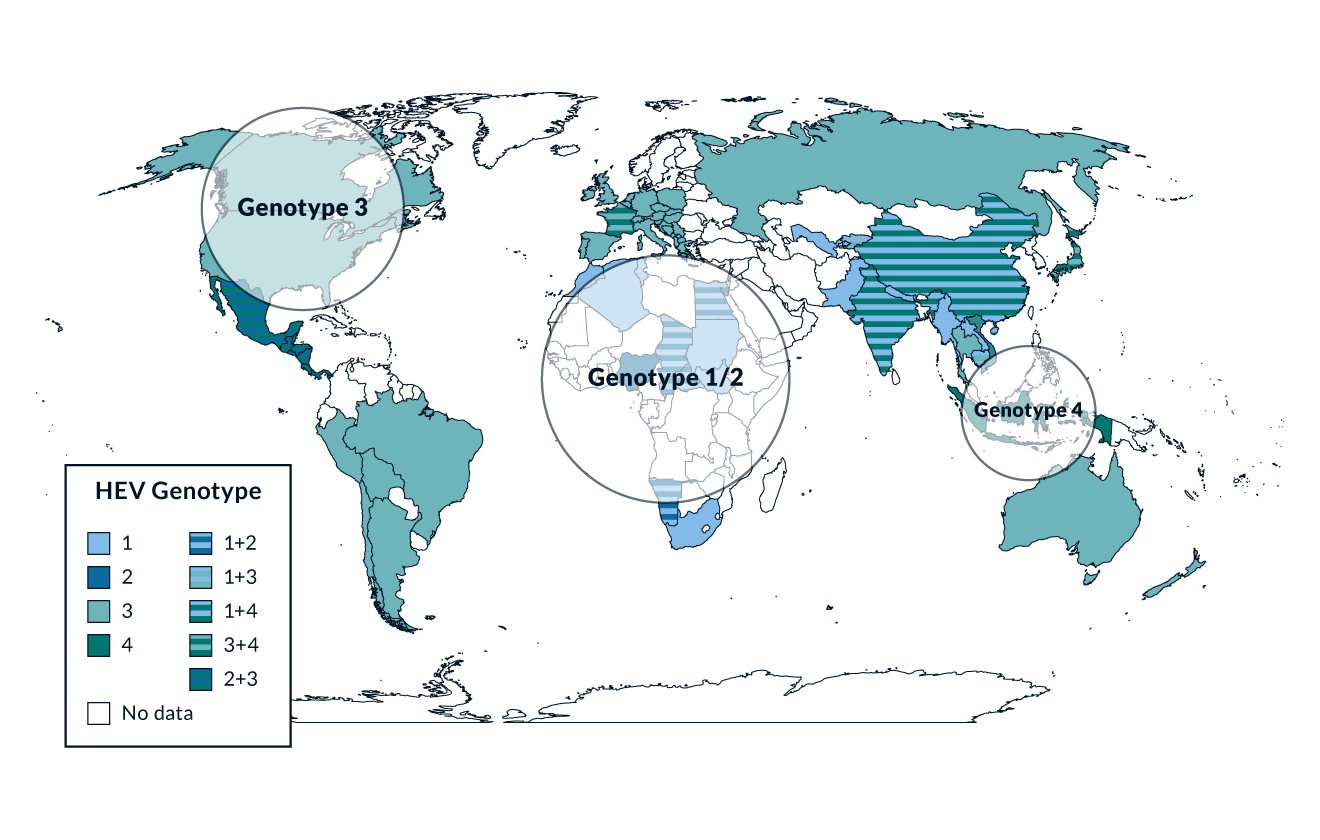 Figure 1. Worldwide distribution of the four main HEV genotypes
Figure 1. Worldwide distribution of the four main HEV genotypes
Patient-to-patient transmission of HEV is very rare but has been described from a large outbreak in Northern Uganda (Teshale 2011) and from hematology wards in Europe (Mansuy 2009). Bloodborne transmission of HEV was suggested in the late nineties (Fainboim 1999). Subsequent studies from Hong Kong, Japan, Great Britain and France confirmed blood transfusions as a possible source of HEV transmission (Wedemeyer 2012). A large study from Germany investigating 1019 blood donors determined that 0.35% seroconverted within 1 year (Juhl 2013). A study from the Netherlands revealed that 13 out of 40, 176 blood donors were HEV-viremic (Slot 2013). These data correspond to one HEV positive blood donation per day in the Netherlands. A large study from England investigating 225, 000 blood products confirmed blood transfusions as a possible source for HEV transmission, in that 0.035% of blood products were viremic for HEV (Hewitt 2014). Post-transfusion infections were associated with viral load in the blood product and absence of HEV antibodies. A study from the Netherlands estimated a duration of 68 day of viraemia within apparently healthy blood donors with subclinical HEV infections (Hogema 2015). These observations led to many countries now routinely testing blood donations for HEV RNA to exclude bloodborne transmission of this virus. HEV transmission by solid organ transplantation is possible but rare (Schlosser 2011).
In summary, there are many different sources of infection for HEV transmission (Figure 2).
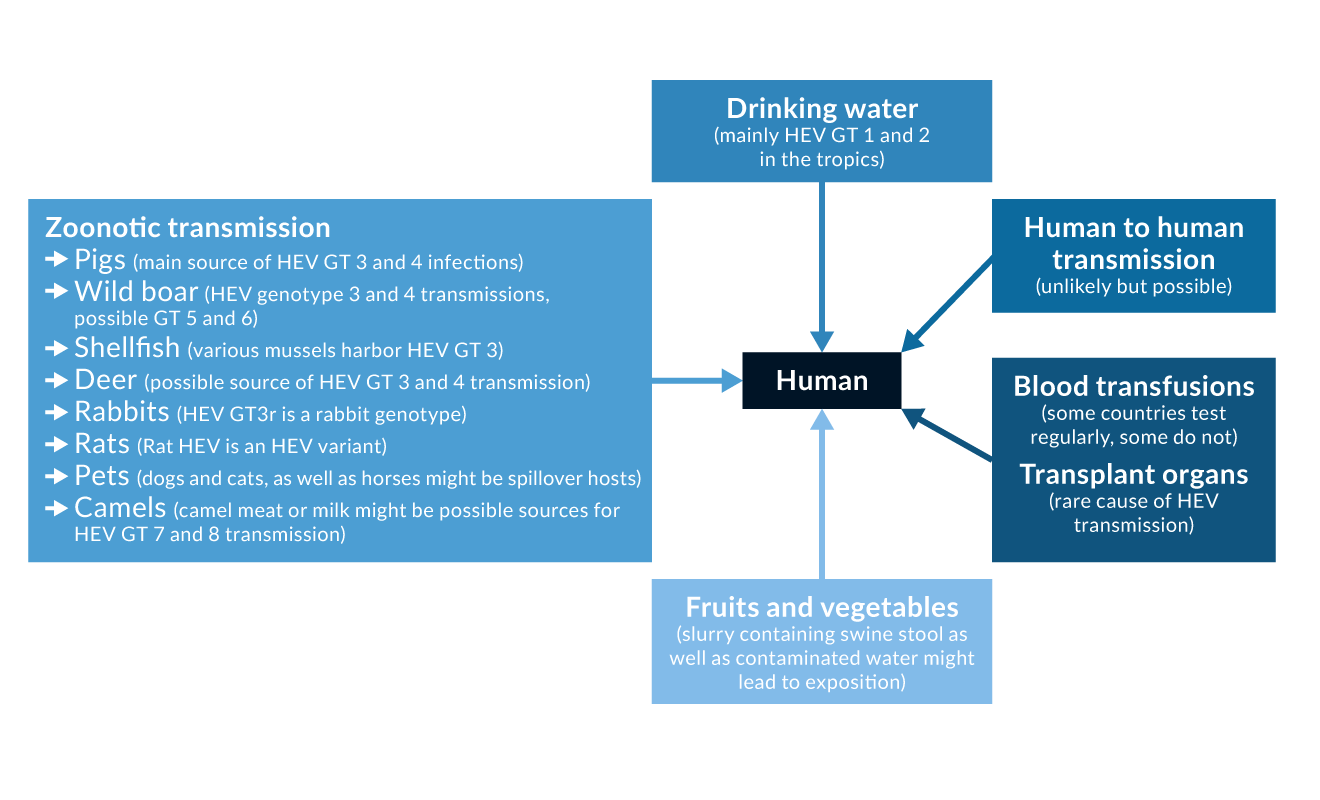 Figure 2. Possible sources of HEV infection
Figure 2. Possible sources of HEV infection
Diagnostic
In immunocompetent patients the diagnosis of hepatitis E usually relies on the detection of HEV-specific antibodies. While IgG antibodies indicate acute and past HEV infections, IgM antibodies can only be found in patients with recent infections (Wedemeyer 2012). There are different commercial assays available for detection of HEV-specific antibodies. Comparison of six of these assays reveals a wide variation of diagnostic sensitivities and specificities as well as interassay disagreements (Drobeniuc 2010). Thus, some of the remarkable discrepancies in HEV seroprevalence rates reported in different studies may be explained by varying sensitivities of the respective assays.
HEV-specific IgG antibodies can be detected in patients with previous contact with HEV. They do not differentiate between ongoing HEV infection and past contact with the virus. To prove current infection the detection of HEV RNA by PCR has been established. Numerous assays using different primers have been developed (Meng 1999, Zhao 2007). Furthermore, few quantitative PCR assays have been described (Ahn 2006, Enouf 2006). Recently a novel WHO-approved RNA standard assay has been developed (Baylis 2011).
In immunocompromised individuals, diagnosis of HEV infection may only be based on the detection of HEV RNA as seroassays lack sensitivity especially in the early phase of infection (Pischke 2010b). HEV RNA can not only be detected in serum samples but also in stool (Wedemeyer 2012), and thus infectivity of HEV infected persons can be determined by investigating stool for HEV RNA. Furthermore HEV could be detected in urine (Geng 2015), but the clinical relevance of this observation still needs to be determined. An HEV antigen assay for detection of HEV has been described (Gupta 2013) but the clinical value of this test still needs to be verified.
Acute hepatitis E in immunocompetent individuals
In the vast majority of cases, contact with HEV takes an asymptomatic course (Stoszek 2006, Wedemeyer 2012, Wedemeyer 2013), especially if the contact happens during childhood (Buti 2008). Immunocompetent individuals should be able to clear the virus spontaneously. In symptomatic cases the incubation period of HEV infections ranges from three to eight weeks with a mean of 40 days (Wedemeyer 2012). The peak of HEV viraemia can be detected in the early phase of infection while the peak of ALT elevation usually occurs around 6 weeks after infection (Wedemeyer 2012).
Initial symptoms in acute hepatitis E are typically unspecific and can include flu-like myalgia, arthralgia, weakness and vomiting. In some patients jaundice, itching, uncoloured stool and darkened urine occur accompanied by elevation of liver transaminases, bilirubin, alkaline phosphatase and gamma-glutamyltransferase.
HEV infection can lead to more severe acute liver disease in pregnant women or patients with underlying chronic liver diseases progressing to fulminant hepatic failure in individual cases (Wedemeyer 2012). Possible explanations for the more severe course in pregnant women are hormonal and immunological changes during pregnancy, they are also associated with the genotype 1 (Navaneethan 2008). Recently, an association between reduced expression of the progesterone receptor and fatal outcome of hepatitis E in pregnant women has been reported (Bose 2011).
Single cases of prolonged courses of HEV infection in immunocompetent individuals with up to two years of viraemia have been described from France (Mallet 2010), Spain (Gonzalez Tallon 2011) and China (Liu 2011). However, no case of HEV-associated liver cirrhosis or development of hepatocellular carcinoma has been reported in immunocompetent individuals. Prolonged HEV viraemia may indicate a previously undiagnosed disturbance of the immune system in otherwise healthy individuals (Höner zu Siederdissen 2014).
Acute and chronic HEV infections in organ transplant recipients
Chronic courses of HEV infection have been described in European liver or kidney transplant recipients since 2008 (Gerolami 2008, Haagsma 2009, Kamar 2008, Pischke 2010b, Behrendt 2014). 14 cases of hepatitis E were initially reported in kidney- and liver-transplanted patients from southwest France (Kamar 2008). Eight of them developed a chronic course leading to persistently elevated ALT levels and significant inflammatory activity and fibrosis in liver biopsies after a follow-up of more than 12 months (range 10 to 18). Subsequently, additional cases of chronic HEV infections have been reported in transplant patients by several groups (Wedemeyer 2012), clearly demonstrating that chronic hepatitis E can be associated with progressive liver disease in patients after organ transplantation (Kamar 2011c).
A study from Germany examined 226 liver-transplanted patients and 129 patients with chronic liver disease to evaluate the frequency of chronic HEV infections in liver transplant recipients in a low endemic country (Pischke 2010b). All patients were tested for HEV RNA and anti-HEV IgG. Two cases of chronic HEV infections in liver transplanted patients were identified. One of them developed significant liver fibrosis (ISHAK F3) within less than 2 years. Both patients were infected with HEV genotype 3. The possibility of reverse zoonotic transmission was experimentally confirmed by infecting pigs with a patient’s blood. HEV RNA was detectable in various organs of the pigs including muscle. Thus, these findings further support the recommendations that eating uncooked meat should be avoided by organ transplant recipients as this may represent a source for acquiring HEV infection.
Retrospective data on hepatitis E in transplant recipients were summarised from 17 centres. Overall, 85 cases of HEV infection were described, 56 (66%) of whom developed chronic hepatitis E. Of note, chronicity was associated with the use of tacrolimus and with low platelet count (Kamar 2011c). However it has to be considered that the vast majority of patients had been recruited by one centre and experiences from other regions and transplant centres need to be reported.
Chronic courses of HEV infection have also been reported in heart transplant recipients (de Man 2011, Pischke 2012b). A study from Germany investigating heart transplant recipients and non-transplant cardiac patients revealed that the seroprevalence of HEV-specific antibodies is increased 5-fold in these patient groups in comparison to healthy controls (Pischke 2012b). It has been assumed that medical procedures, especially blood products, could explain this difference in seroprevalence rates.
Chronic HEV infections have also been described in lung transplant recipients from the Netherlands (Rizebos-Brilman 2013) and Germany (Pischke 2014).
Overall, all recipients of solid organ transplant with elevated liver enzymes should be tested for HEV RNA unless other obvious reasons already explain the hepatitis. In immunosuppressed patients, testing for HEV RNA should be applied as antibody testing may lack sensitivity. Distinct immunosuppressive drugs may indirectly or directly effect HEV replication, which needs to be considered in the management of organ transplant recipients (Behrendt 2014).
In contrast to solid organ transplant recipients, studies from Germany (Koenecke 2012) and France (Abravanel 2012) did not observe any case of chronicity in stem cell transplant recipients, leading to the assumption that this phenomenon is rare in this patient population. However, a large study from the Netherlands, investigating 328 stem cell transplant recipients, identified 8 cases (2.4%) of chronic HEV viraemia. Four of these patients died after development of hepatitis, while the other four patients cleared HEV infection after a median period of 6.3 months. These data demonstrate that chronic HEV infections in stem cell transplant recipients are indeed relevant (Versluis 2013).
Hepatitis E in patients with HIV infection or other immunological deficiencies
Chronic hepatitis E was described for the first time in a patient with underlying HIV infection in 2009 (Dalton 2009). This patient had a CD4 T cell count of less than 200 cells and high HIV RNA levels (>100, 000 copies/mL). However, subsequent studies from Spain (n=93) (Madejon 2009), Germany (n=123) (Pischke 2010a) and England (n=138) (Keane 2012) could not identify cases of chronic hepatitis E in HIV-infected individuals. HEV RNA was detected for more than 10 months in only one out of 184 HIV positive individuals in France (Kaba 2010). This patient had particularly low CD4 counts (<50 cells/mm) while two additional patients with higher CD4 levels were able to clear HEV spontaneously. Thus, persistent HEV infection is rarely observed in HIV-infected patients. Recently, it has been demonstrated that HEV may persist in HIV infected patients despite improvement of their immune system (Kuniholm 2015).
In addition to HIV infected patients, chronic HEV infections in patients with different underlying conditions of immunosuppression including lupus erythematodes, granulomatosis, retroperitoneal fibrosis or CD4 deficiency have been reported (Grewal 2013, Höner zu Siederdissen 2014). In contrast to these diseases, there was no case of chronic HEV infection within a German cohort of 73 patients with common variable immunodeficiency (CVID). It has been hypothesised, that eventually regular immunoglobulin infusions in these patients may have protected them from infection (Pischke 2012a).
Extrahepatic manifestations of hepatitis E
There is some evidence that HEV infections are associated with extrahepatic manifestations, particularly neurological, immunological and renal diesases. Neurological symptoms associated with acute or chronic HEV infection have been described in single cases in the past few few years (Kamar 2011b). More recently, HEV infections were linked with neuralgic amyotrophy (van Eijk 2014), Guillain-Barré syndrome (Van den Berg 2014) and common inflammatory demyelinisating polyneuropathy (Pischke 2024). The underlying mechanisms and the clinical relevance of these associations require further investigation.
It still needs to be determined if extrahepatic manifestations are caused by direct effects of the virus or by indirect, immunological mechanisms. A possible link between HEV and cryoglobulinemia has recently been suggested (Pischke 2014, Kamar 2012).
Recently a pathophysiological link between HEV infections and glomerulonephritis has been suggested by demonstrating an association between production of HEV ORF2 protein and the development of glomerulonephritis in a kidney transplant recipient (Leblond 2024).
Therapy and prevention
Treatment options for chronic hepatitis E include reduction of immunosuppression, administration of pegylated interferon α or use of ribavirin. The first step in the treatment of chronic HEV infection should be to evaluate if it is possible to reduce the immunosuppressive medication (Wedemeyer 2012). Reduction of immunosupression in 16 solid organ transplant recipients with chronic hepatitis E led to clearance of HEV in 4 cases (25%) (Kamar 2011a). A second possible treatment option is the use of PEG-IFN α (Haagsma 2010, Kamar 2010a). Treatment durations varied between 3 and 12 months. Overall, 4 out 5 patients were successfully treated with sustained clearance of HEV RNA. However, the use of interferon can be associated with significant side effects and may cause rejection in organ transplant recipients. Interferon α is therefore not recommended in heart or kidney transplant recipients. Another therapeutic option for HEV infections is the off-label use of Ribavirin. The antiviral efficacy of ribavirin monotherapy has been evaluated by two French groups (Kamar 2010b, Mallet 2010). A sustained virological response was observed in 2/2 and 4/6 treated patients, respectively. Ribavirin has also been used in a non-transplanted patient with severe acute hepatitis E who showed rapid improvement of symptoms and liver function tests during treatment (Gerolami 2011).
A study from France demonstrated the safe use of ribavirin in non transplant individuals with acute HEV genotype 3 infections (Peron 2015). Furthermore the use of ribavirin has been demonstrated in one single case with severe HEV genotype 1 infection (Pischke 2013a). Starting and stopping rules for the treatment of HEV with ribavirin still need to be defined. In contrast to immunocompetent individuals, in solid organ transplant recipients with chronic HEV infection ribavirin remains a frequently used therapeutic option. A multicentric French study confirmed that treatment of chronic HEV infections in transplant recipients with ribavirin is safe and efficient (Kamar 2014). However, ribavirin treatment failures have been described in single patients (Pischke 2012b, Pischke 2013a) that may be linked to selection of a distinct HEV polymerase variant with increased replication fitness (Debing 2014).
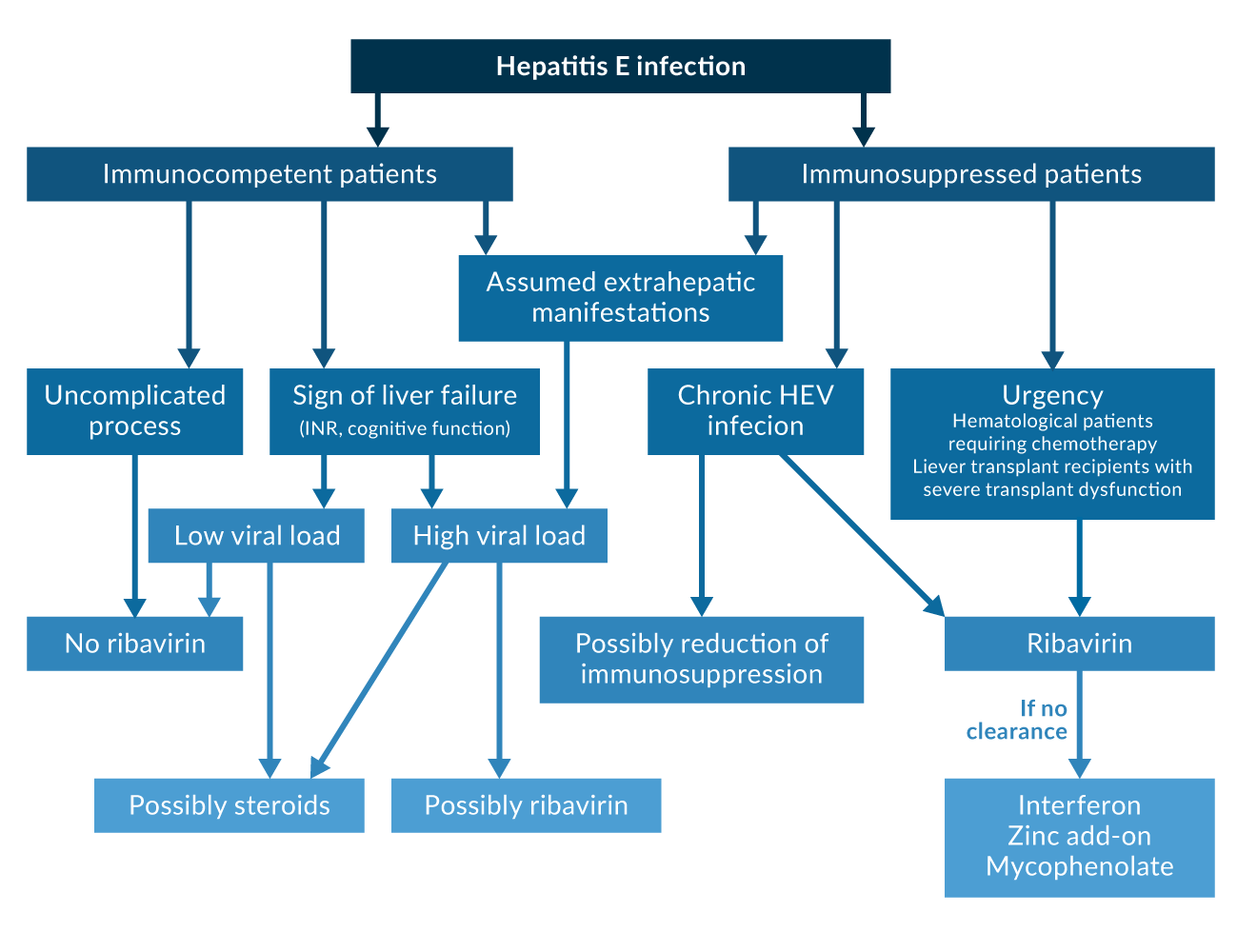 Figure 3. Treatment algorythm
Figure 3. Treatment algorythm
Sofosbuvir, as a novel antiviral approach, failed to achieve HEV RNA elimination in a pilot study of 9 patients (Cornberg 2020). Recently, distinct resistance mutations in the RNA dependent RNA polymerase were identified in this patient cohort (Gömer 2023). The therapeutic effect of a combinational therapy of Sofosbuvir and Ribavirin has yet to be investigated in bigger cohorts.
In addition to the treatment of acute or chronic HEV infection, the possibility of preventing a relevant infection in the event of HEV exposure through vaccination is also of great importance.
A vaccine developed by GSK and the Walter Reed Army Institute that was successfully tested in a phase 2 study (Shrestha 2007) has not been further developed. A group from China reported data from a very large successful phase 3 vaccine trial (Zhu 2010). This trial included almost 110, 000 individuals who received either a recombinant HEV vaccine (“Hecolin”) or placebo. The vaccine efficacy after three doses was 100%. This vaccine was approved in China in early 2012 and in Pakistan in 2020. It is currently not known if and when this vaccine will become available elsewhere. Moreover, the efficacy of this vaccine needs to be evaluated in special risks groups such as patients with end-stage liver disease or immunosuppressed individuals. It is also unknown if HEV 239 also protects from HEV genotype 3 infection (Wedemeyer 2011). However, it was demonstrated that either the vaccine or naturally acquired, post-infectious antibodies are able to prevent symptomatic hepatitis E, but not asymptomatic infection (Zhang 2013). Furthermore, it was shown that this vaccine could be safely used in pregnant women (Wu 2012). In contrast to this study, a recent double-blind placebo-controlled trial from Bangladesh showed containing 5011 pregnant women vaccinated prior to pregnancy showed a significantly increased risk of the group of women receiving HEV vaccine for miscarriage (Binte-Aziz 2024).
Furthermore a follow-up study of the initial Hecolin vaccine trial demonstrated a 10-year efficacy above 80% (Huang 2024).
The use of this vaccine in developing countries needs to be discussed and investigated. Eventually this vaccine may help to prevent the morbidity and mortality caused by hepatitis E.
Conclusions and recommendations
In general, HEV infection has a self-limiting course associated with the clinical picture of acute hepatitis in immunocompetent populations. Special populations like pregnant women may be more likely to develop hepatic failure. In patients with immunosuppression of different aetiologies, chronic cases have been reported.
In organ transplant recipients, the diagnosis of HEV infection should not be based on serological assays alone as these assays may lack sensitivity. Detection of HEV RNA by PCR in serum or stool represents the gold standard for diagnosis of HEV infection.
The prevalence of chronic HEV infection in solid organ transplant recipients depends on the general prevalence in the population and is low in most industrialised countries. However, chronic hepatitis E occurs and needs to be considered in the differential diagnosis of graft hepatitis, as persistent HEV infection can be associated with progressive graft hepatitis and the development of liver cirrhosis. Currently, all reported cases of chronic HEV infections in transplant recipients have been due to HEV genotype 3 or 4. It is not known if chronic hepatitis E can also be caused by the genotypes 1 or 2.
Organ transplant recipients and other immunocompromised individuals should be made aware of this risk and avoid eating uncooked meats.
First results indicate that ribavirin treatment of chronic hepatitis E (3 to 5 months duration) is effective to achieve sustained virological response in immunocompromised persons. In contrast, in immunocompetent individuals with acute HEV infection this treatment is only required in few cases to avoid liver failure.
Due to the side effects of the current available antiviral therapies as well as the many cases of relapse after therapy, novel treatment options for chronic HEV infections need to be investigated. Additionally, an optimisation of the evaluation of therapeutic response might be possible by investigating HEV antigen in urine.
A widely available and efficacious vaccine would prove important to lower the overall burden HEV infections cause on the health system.
The relevance of extrahepatic manifestations associated with acute or chronic HEV infection needs further exploration, especially the association between positive anti-HEV serostatus and autoimmune hepatitis, cryoglobulinaemia or neurological symptoms.
Key Messages
- HEV is the most prevalent causative agent of acute viral hepatitis worldwide
- Most infections are asymptomatic and self-resolving, pregnant women and people with underlying liver disease are at risk of fulminant hepatitis
- Immunosuppressed individuals, especially organ transplant recipients, are at risk of chronic infection, leading to liver fibrosis and cirrhosis
- In organ transplant recipients with unclear elevation of liver enzymes, HEV infection should be ruled out by PCR testing
References
Abravanel F, Mansuy JM, Huynh A, Kamar N, Alric L, Peron JM, et al. Low risk of hepatitis E virus reactivation after haematopoietic stem cell transplantation. J Clin Virol. 2012;152-5.
Ahn JM, Rayamajhi N, Gyun Kang S, Sang Yoo H. Comparison of real-time reverse transcriptase-polymerase chain reaction and nested or commercial reverse transcriptase-polymerase chain reaction for the detection of hepatitis E virus particle in human serum. Diagn Microbiol Infect Dis 2006;56:269-74.
Baylis S, Hanschmann KM, Blümel J, Nübling CM; HEV Collaborative Study Group. Standardisation of hepatitis E virus (HEV) nucleic acid amplification technique-based assays: an initial study to evaluate a panel of HEV strains and investigate laboratory performance. J Clin Microbiol. 2011;1234-9.
Behrendt P, Steinmann E, Manns MP, Wedemeyer H. The impact of hepatitis E in the liver transplant setting. J. Hepatol. 2014;61:1418-1429.
Binte Aziz A,, Dudman S, Halle Julin CH, et al. Receipt of hepatitis E vaccine and fetal loss in rural Bangladesh: further analysis of a double-blind, cluster-randomised, controlled trial. Lancet Global Health 2024 Aug;12(8):e1300-e1311
Bose PD, Das BC, Kumar A, Gondal R, Kumar D, Kar P. High viral load and deregulation of the progesterone receptor signaling pathway: Association with Hepatitis E-related poor pregnancy outcome. J Hepatol 2011;54:1107-13.
Buti M, Plans P, Dominguez A, et al. Prevalence of hepatitis E virus infection in children in the northeast of Spain. Clin Vaccine Immunol 2008;15:732-4.
Caron M, Enouf V, Than SC, Dellamonica L, Buisson Y, Nicand E. Identification of genotype 1 hepatitis E virus in samples from swine in Cambodia. J Clin Microbiol. 2006;3440-2.
CDC, Investigation of hepatitis E outbreak among refugees - Upper Nile, South Sudan, 2012-2013. MMWR Morb Mortal Wkly Rep. 2013;26:581-6.
Clemente-Casares P, Pina S, Buti M, et al. Hepatitis E virus epidemiology in industrialised countries. Emerg Infect Dis 2003;9:448-54.
Colson P, Borentain P, Queyriaux B, et al. Pig liver sausage as a source of hepatitis E virus transmission to humans. J Infect Dis 2010;202:825-34.
Cornberg M, Pischke S, Müller T, et al. Sofosbuvir monotherapy fails to achieve HEV RNA elimination in patients with chronic hepatitis E - The HepNet SofE pilot study. J Hepatol. 2020 Sep;73(3):696-699.
Dalton HR, Bendall RP, Keane FE, Tedder RS, Ijaz S. Persistent carriage of hepatitis E virus in patients with HIV infection. N Engl J Med 2009;361:1025-7.
Debing Y, Gisa A, Dallmeier K, et al. Ribavirin treatment failure in chronic hepatitis E is associated with an enhanced fitness polymerase variant. Gastroenterology 2014;147:1008-1011.
de Man RA, Pas SD, Osterhaus AD, Balk AH, Van der Eijk A. Diagnosis and clinical consequences of hepatitis E virus infection in orthotopic heart transplant recipients. Hepatology 2011; AASLD annual meeting.
Drobeniuc J, Meng J, Reuter G, et al. Serologic assays specific to immunoglobulin M antibodies against hepatitis E virus: pangenotypic evaluation of performances. Clin Infect Dis 2010;51:e24-7.
Emerson SU, Arankalle VA, Purcell RH. Thermal stability of hepatitis E virus. J Infect Dis 2005;192:930-3.
Enouf V, Dos Reis G, Guthmann JP, et al. Validation of single real-time TaqMan PCR assay for the detection and quantitation of four major genotypes of hepatitis E virus in clinical specimens. J Med Virol 2006;78:1076-82.
Fainboim H, Gonzalez J, Fassio E, et al. Prevalence of hepatitis viruses in an anti-human immunodeficiency virus positive population from Argentina. A multicentre study. J Viral Hepat 1999;6:53-7.
Geng Y, Wang C, Zhao C, et al. Serological Prevalence of Hepatitis E Virus in Domestic Animals and Diversity of Genotype 4 Hepatitis E Virus in China. Vector Borne Zoonotic Dis 2009.
Geng Y, Zhao C, Huang W, et al. Detection and assessment of infectivity of hepatitis E virus in urine. J Hepatol 2016; 64(1):37-43
Gerolami R, Borentain P, Raissouni F, Motte A, Solas C, Colson P. Treatment of severe acute hepatitis E by ribavirin. J Clin Virol 2011;52:60-2.
Gerolami R, Moal V, Colson P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N Engl J Med 2008;358:859-60.
Gonzalez Tallon AI, Moreira Vicente V, Mateos Lindemann ML, Achecar Justo LM. [Chronic hepatitis E in an immunocompetent patient.]. Gastroenterol Hepatol 2011.
Gömer A, Klöhn M, Jagst M, Nocke M, Pischke S, Horvatits T, Schulze Zur Wiesch J, Müller T, Hardtke S, Cornberg M, Wedemeyer H, Behrendt P, Steinmann E, Todt D. Emergence of resistance-associated variants during sofosbuvir treatment in chronically infected hepatitis E patients. Hepatology. 2023 Jun 20.
Grewal P, Kamili S, Motamed D. Chronic hepatitis E in an immunocompetent patient: A case report. Hepatology 2014;59:347-8.
Gupta E, Pandey P, Sharma MK, et al. Role of Hepatitis E virus antigen in confirming active viral replication in patients with acute viral hepatitis Einfection. J. Clin. Virology 2013;58; 374-377.
Haagsma E, Riezebos-Brilman A, Van den Berg AP, Porte RJ, Niesters HG. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transpl 2010. [epub ahead of print]
Haagsma EB, Niesters HG, van den Berg AP, et al. Prevalence of hepatitis E virus infection in liver transplant recipients. Liver Transpl 2009;15:1225-8.
Hakze-van der Honing RW, van Coillie E, Antonis AF, van der Poel WH. First Isolation of Hepatitis E Virus Genotype 4 in Europe through Swine Surveillance in the Netherlands and Belgium. PLoS One 2011;6:e22673.
Helsen N, Debing Y, Paeshuyse J, et al. Stem cell derived hepatocytes: a novel model for hepatitis E virus replication. J Hepatol 2015, pii: S0168-8278(15)00771-0, epub ahead of print
Hewitt PE, Ijaz S, Braisford SR, et al. Hepatitis E virus in blood components: a prevalence and transmission study in southeast England. Lancet 2014;1766-1773.
Höner zu Siederdissen C, Pischke S, Schlue J, et al. Chronic HEV infection beyond transplantation or HIV infection. Hepatology 2014;60; 1112-1113.
Hogema BM, Molier M, Sjerps M, et al. Incidence and duration of hepatitis E virus infections in Dutch blood donors. Transfusion 2015, epub ahead of print.
Huang S, ZhangX, Su Y, et al. Long-term efficacy of a recombinant hepatitis E vaccine in adults: 10-year results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2024; Mar 2;403(10429):813-823.
Juhl D, Baylis SA, Blumel J, Gorg S, Hennig H. Seroprevalence and incidence of hepatitis E virus infection in German blood donors. Transfusion 2013; 54: 49-56.
Kaba M, Richet H, Ravaux I, et al. Hepatitis E virus infection in patients infected with the human immunodeficiency virus. J Med Virol 2010;83:1704-16.
Kamar N, Abravanel F, Selves J, et al. Influence of immunosuppressive therapy on the natural history of genotype 3 hepatitis-E virus infection after organ transplantation. Transplantation 2011a;89:353-60.
Kamar N, Bendall RP, Peron JM, et al. Hepatitis E virus and neurologic disorders. Emerg Infect Dis 2011b;17:173-9.
Kamar N, Garrouste C, Haagsma EB, et al. Factors associated with chronic hepatitis in patients with hepatitis e virus infection who have received solid organ transplants. Gastroenterology 2011c;140:1481-9.
Kamar N, Izopet J, Cintas P, et al. Hepatitis e virus-induced neurological symptoms in a kidney-transplant patient with chronic hepatitis. Am J Transplant 2011d;10:1321-4.
Kamar N, Rostaing L, Abravanel F, et al. Pegylated Interferon-alpha for Treating Chronic Hepatitis E Virus Infection after Liver Transplantation. Clin Infect Dis 2010a.
Kamar N, Rostaing L, Abravanel F, et al. Ribavirin Therapy Inhibits Viral Replication on Patients With Chronic Hepatitis E Virus Infection. Gastroenterology 2010b.
Kamar N, Selves J, Mansuy JM, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 2008;358:811-7.
Kamar N, Izopet J, Tripon S, et al. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N. Engl. J. Med. 2014; 370, 1111-1120
Kamar N, Weclawiak H, Guilbeau-Frugier C, et al.. Hepatittis E Virus and the kidney in Solid-Organ transplant patients. Transplantation 2012;6: 617-623.
Keane F, Gompels M, Bendall R, et al. Hepatitis E virus coinfection in patients with HIV infection. HIV Med 2012 Jan;13(1):83-88.
Koenecke C, Pischke S, Heim A, Raggub L, Bremer B, Raupach R, et al. Chronic hepatitis E in hematopoietic stem cell transplant patients in a low-endemic country? Transpl Infect Dis. 2012:103-6.
Kuniholm MH, OngE, Hogema BM, et al. Acute and Chronic Hepatitis E Virus Infection in HIV-infected United States Women. Hepatology 2015. Epub ahead of print
Leblond AL, Helmchen B, Ankavay M, et al. HEV ORF2 protein-antibody complex deposits are associated with glomerulonephritis in hepatitis E with reduced immune status. Nat Commun. 2024 Oct 14;15(1):8849
Lee GH, Tan BH, Teo EC, et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver-transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2015 epub ahead of print.
Liu L, Liu Y. Analysis of acute to chronic hepatitis E: 6-10 year follow-up. Hepatogastroenterology 2011;58:324-5.
Madejon A, Vispo E, Bottecchia M, Sanchez-Carrillo M, Garcia-Samaniego J, Soriano V. Lack of hepatitis E virus infection in HIV patients with advanced immunodeficiency or idiopathic liver enzyme elevations. J Viral Hepat 2009.
Mallet V, Louvet A, Chakvetadze C, et al. Ribavirin Treatment for Chronic Hepatitis E: a Case-Series. Hepatology 2010;52:919A-1020A.
Melenhorst WB, Gu YL, Jaspers WJ, Verhage AH. Locally acquired hepatitis E in the Netherlands: associated with the consumption of raw pig meat? Scand J Infect Dis 2007;39:454-6.
Meng J, Cong M, Dai X, et al. Primary structure of open reading frame 2 and 3 of the hepatitis E virus isolated from Morocco. J Med Virol 1999;57:126-33.
Navaneethan U, Al Mohajer M, Shata MT. Hepatitis E and pregnancy: understanding the pathogenesis. Liver Int 2008;28:1190-9.
Peron JM, Abravanel F, Guillaume M, et al.Liver International 2015, epub ahead of print
Pischke S, Heim A, Bremer B, et al. Hepatitis E: An Emerging Infectious Disease in Germany? Z Gastroenterol 2011a;49:1255-1257.
Pischke S, Hardtke S, Bode U, et al. Ribavirin treatment of acute and chronic hepatitis E: a single center experience. Liver International 2013a;33:722-6.
Pischke S, Geer M, Hardtke S, et al. Course and treatment of chronic hepatitis E virus infection in lung transplant recipients. Transpl. Inf. Dis. 2014;16, 333-339
Pischke S, Gisa A, Suneetha PV, et al. Increased HEV seroprevalence in patients with autoimmune hepatitis. PLOS One 2014, e85330.
Pischke S, Ho H, Urbanek F, et al. Hepatitis E in HIV positive patients in a low-endemic country. J Viral Hepat 2010a;17:598-9.
Pischke S, Horn-Wichmann R, Ernst, D, et al. Absence of chronic hepatitis E in a German cohort of common variable immunodeficiency patients. Infectious Disease Reports 2012a;volume 4:e28.
Pischke S, Kjasimov A, Skripuletz T, et al. Serological indication of chronic inflammatory demyelinating polyneuropathy as an extrahepatic manifestation of hepatitis E virus infection. Scientific reports 2024 Aug 20;14(1):19244.
Pischke S, Stiefel P, Franz B, Bremer B, Suneetha PV, Heim A, et al. Chronic hepatitis e in heart transplant recipients. Am J Transplant. 2012b:3128-33.
Pischke S, Suneetha PV, Baechlein C, et al. Hepatitis E virus infection as a cause of graft hepatitis in liver transplant recipients. Liver Transpl 2010b;16:74-82.
Pischke S, Voigtlaender T, Koenecke C, et al. Chronic hepatitis E in immunosuppressed patients beyond liver- and kidney transpantation. Journal of Hepatology 2011b;54, S1:495.
Pischke S, Wedemeyer H. Hepatitis E Virus Infection: multiple faces of an underestimated problem. J. Hepoatol. 2013b;58: 1045-6.
Rein DB, Stevens G, Theaker J, Wittenborn JS, Wiersma ST. The global burden of hepatitis E virus. Hepatology 2011;55:988-97.
Rizebos-Brilman A, Puchhammer-Stockl E, Van der Weide HY, et al. Chronic hepatitis E infection in lung transplant recipients. J Heart Lung Transplant 2013;32:341-6.
Rivero-Juarez A, Frias M, Perez AB, Pineda JA, et al. Orthohepevirus C infection as an emerging cause of acute hepatitis in Spain: First report in Europe. J Hepatol. 2022 Aug;77(2):326-331.
Schlosser B, Stein A, Neuhaus R, et al. Liver transplant from a donor with occult HEV infection induced chronic hepatitis and cirrhosis in the recipient. J Hepatol 2011.
Shrestha MP, Scott RM, Joshi DM, et al. Safety and efficacy of a recombinant hepatitis E vaccine. N Engl J Med 2007;356:895-903.
Slot E, Hogema BM, Rizebos-Brillman A, Kok TM, Molier M, Zaaijer HL. Silent hepatitis E virus infection in Dutch blood donors, 2011 to 2012. Euro Surveill. 2013;18 pii: 20550.
Smith DB, Simmonds P, Izopet J, et al. J Gen Virol 2016, epub ahead of print
Stoszek SK, Engle RE, Abdel-Hamid M, et al. Hepatitis E antibody seroconversion without disease in highly endemic rural Egyptian communities. Trans R Soc Trop Med Hyg 2006;100:89-94.
Teshale EH, Grytdal SP, Howard C, et al. Evidence of person-to-person transmission of hepatitis E virus during a large outbreak in Northern Uganda. Clin Infect Dis 2011;50:1006-10.
Van den Berg B, Annemick A, Van den Eijk A, et al. Guillain-Barré syndrome associated with preceding hepatitis E virus infection. Neurology 2013;82, 491-497.
Van Eijk JJ, Madden RG, van der Eijk AA, et al. Neuralgic amyotrophy and hepatitis E virus infection. Neurology 2014;82, 498-503.
Versluis J, Pas SD, Agteresch HJ, et al. Hepatitis E virus: an underestimated opportunistic pathogen in recipients of allogeneic hematopoietic stem cell transplantation. Blood 2013;122:1079-86.
Wedemeyer H, Pischke S, Manns MP. Pathogenesis and treatment of hepatitis e virus infection. Gastroenterology 2012;1388-1397.
Wedemeyer H, Pischke S. Hepatitis: Hepatitis E vaccination-is HEV 239 the breakthrough? Nat Rev Gastroenterol Hepatol 2011;8:8-10.
Wedemeyer, H, Rybczynska J, Pischke S, Krawczynski K. Immunopathogenesis of hepatitis E virus infection. Semin. Liver Dis. 2013;33: 71-78.
Wenzel JJ, Preiss J, Schemmerer M, Huber B, Plentz A, Jilg W. Detection of hepatitis E virus (HEV) from porcine livers in Southeastern Germany and high sequence homology to human HEV isolates. J Clin Virol 2011.
WHO, Hepatitis E Factsheet No 280, Updated June 2014, http://www.who.int/mediacentre/factsheets/fs280/en [Accessed 20 January 2015]
WHO, Acute hepatitis E – South Sudan, updated 5 May 2023, https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON466 [accessed 12 October 2023]
WuT, Zhu FC, Huang SJ, et al. Safety of the hepatitis E vaccine for pregnant women: A preliminary analysis. Hepatology 2012;55: 2038.
Zhang J, Zhang XF, Zhou C, et al. Protection against hepatitis E virus infection by naturally acquired and vaccine-induced immunity. Clin Microbiol Infect 2013; epub ahead of print.
Zhao ZY, Ruan B, Shao H, Chen ZJ, Liu SL. Detection of hepatitis E virus RNA in sera of patients with hepatitis E by polymerase chain reaction. Hepatobiliary Pancreat Dis Int 2007;6:38-42.
Zhu FC, Zhang J, Zhang XF, et al. Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: a large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 2010;376:895-902.